Les méthodes de machine learning (ML) ont fait récemment leur apparition dans le domaine des simulations atomistiques. Dans leur principe, elles offrent la possibilité d’obtenir rapidement un résultat assez précis pour des grandeurs physiques comme l’énergie ou les forces interatomiques pour un ensemble d’atomes. L’enjeu est bien sûr de simuler beaucoup plus rapidement des phénomènes complexes ou des ensembles d’atomes plus grands. Le coût numérique est reporté sur la phase d’apprentissage permettant de paramétrer le modèle à l’aide de calculs de référence. Si l’approche apparait fructueuse, elle suppose cependant un certain renoncement dans la connaissance des détails, l’impossibilité d’améliorer le résultat par approximations successives et l’impossibilité de transférer le paramétrage à un matériau différent sans refaire intégralement le cycle d’apprentissage.
Dans le cadre de la thèse de Paul Guibourg réalisée au CIMAP, une telle approche a été développée, afin de simuler l’émission d’ions par effet de champ issus d’agrégats chargés de SiC. Ce travail s’inscrit dans le cadre de la collaboration du CIMAP avec le GPM de Rouen pour étudier ce phénomène d’émission par effet de champ, qui constitue le principe de fonctionnement de la sonde atomique tomographique développée par ce laboratoire. L’originalité de l’approche se situe dans l’articulation entre le modèle physique et l’utilisation de Machine Learning pour optimiser le le calcul de façon efficace.
Le Groupe de Physique des Matériaux – GPM de Rouen développe depuis plusieurs décennies une Sonde Atomique Tomographique performante, qui repose sur le phénomène d’émission d’ions par effet de champ. L’instrument permet de faire de l’imagerie 3D avec analyse chimique à l’échelle nanométrique. En collaboration étroite avec ce laboratoire, l’équipe SIMUL du CIMAP étudie le phénomène en apportant un effort de modélisation, qui vise à mieux comprendre la perturbation de la surface induite par l’intense champ électrique appliqué, et relier cette perturbation aux biais de composition souvent observés dans les matériaux semi-conducteurs.
Il existe peu de codes permettant de simuler des réarrangements structuraux de surface en présence de champs électrique fort. Il a donc été nécessaire de créer l’outil adapté pour cette étude. Le cadre théorique est celui de la fonctionnelle de la densité en liaison forte avec charges auto-cohérentes (SCC-DFTB), qui est une approximation de la DFT adaptée au fait que la charge effective d’un atome s’adapte à son environnement atomique. Les codes existants sont limités par le nombre de cycles auto-cohérents à réaliser pour obtenir la convergence des charges. Le travail a donc consisté à reformuler la méthode DFTB en éliminant complètement ces cycles à l’aide d’une méthode de machine learning (ML) pour aboutir à un nouveau formalisme dénommé ML-DFTB.
La partie ML sert ici à prédire les charges portées par les atomes afin que seul reste à effectuer la partie standard du calcul de structure électronique pour obtenir la densité, l’énergie et les forces agissant sur les atomes. Il est important de noter que l’emploi des charges prédites par ML nécessite une reformulation du modèle DFTB afin d’obtenir un calcul inconditionnellement stable. Comme toutes les méthodes paramétrées, et en particulier les méthode d’IA et ML, l’inférence de charge demande une phase d’entraînement du modèle de prédiction, ce qui nécessite un grand nombre de calculs SCC-DFTB de référence pour des configurations atomiques susceptibles d’être rencontrées dans les simulations ultérieures. Cette phase demande généralement plusieurs jours de calculs, tout en restant à la portée d’un petit cluster de calcul.
Cette démarche a été appliquée à des agrégats de SiC chargés. L’utilisation d’agrégats chargés permet de contrôler le champ de surface simplement à l’aide de la charge portée par l’agrégat en fonction de son rayon. La figure montre le potentiel de surface ressenti par un atome de carbone lorsqu’on l’éloigne de la surface de l’agrégat. À l’infini, on en tire l’énergie de cohésion de l’atome chargé, que l’on peut comparer au calcul de référence pour cette grandeur et vérifier ainsi la qualité du modèle.
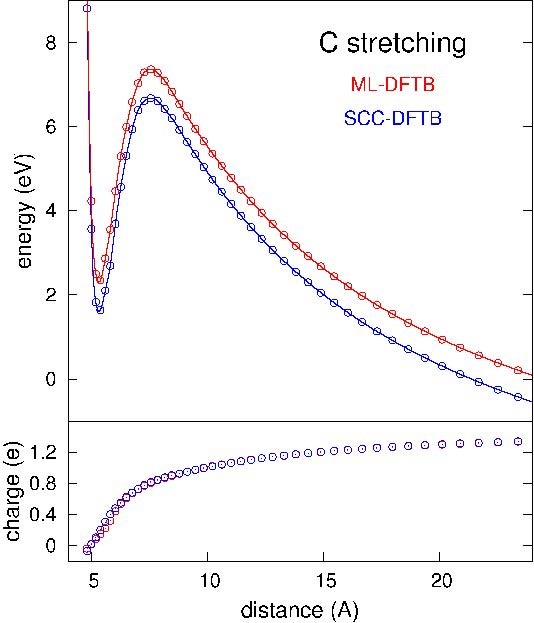
Potentiel et charge de l’ion émis en fonction de la distance à l’agrégat SiC émetteur. Courbe bleue : fonctionnelle de la densité en liaison forte avec charges auto-cohérentes (SCC-DFTB). Courbe rouge : modèle de fonctionnelle de la densité en liaison forte avec Machine Learning (ML- DFTB).
Il reste maintenant à étudier la restructuration de la surface en fonction de la charge portée par l’agrégat, et donc du champ de surface dans la gamme de charge où la barrière d’émission est basse, permettant l’émission d’un atome avec l’apport d’une excitation thermique de faible énergie. Au-delà des études de surface soumises à des champs électriques pour la Sonde Atomique Tomographique, ce travail apporte un corollaire intéressant pour l’étude de la stabilité d’agrégats multi-chargés et fournit un bon exemple de l’apport de méthodes de « machine learning » au calcul DFT.
[1] « DFTB simulation of charged clusters using machine learning charge inference »
Paul Guibourg, Léo Dontot, Pierre-Matthieu Anglade, Benoit Gervais, J Chem Theory Comput 20, 4007-4018 (2024),
Thèse de Paul Guibourg : « Simulatiοn en DFΤB d’agrégats chargés à l’aide de charges οbtenue par méthοde de machine-learning« , soutenue le 23/05/2024 à Caen.
Contact CEA-IRAMIS : B. Gervais, CIMAP/SIMUL
Collaboration :
- Équipe SIMUL du CIMAP, Centre de Recherche sur les Ions, les Matériaux et la Photonique, (UMR CEA-CNRS- Univ. Caen – EnsiCaen), 14050 Caen (France).
- GPM, Groupe de Physique des Matériaux, (UMR 6634, Univ. de Rouen Normandie – INSA de Rouen Normandie – CNRS), Rouen (France).