

Monte Carlo Cinétique pour l'étude de la radio-oxydation |
 |
Sous l’action des rayonnements ionisants en présence d’oxygène, les polymères se dégradent et perdent leurs propriétés d’usage, en particulier leurs propriétés mécaniques. La compréhension des mécanismes de ce vieillissement oxydatif est cruciale pour prédire l’évolution à long terme de ces matériaux. Le modèle actuellement admis a été proposé en 1946 par Bolland et Gee pour la thermo-oxydation des élastomères contenant des insaturations C=C. Il a été graduellement modifié depuis, à l’exception d’une hypothèse majeure : que l’oxydation soit homogène. Or, la structure du dépôt d’énergie par des rayonnements ionisants est par nature hétérogène. Nous avons alors développé une simulation de cinétique d’oxydation basée sur le suivi des radicaux P ⚈ et POO ⚈ , de O2 et des produits d’oxydation résultants (POOH et POOP). Le modèle s’appuie sur des données de diffusion pour les polymères et permet d’étudier la radio-oxydation par des ions, comparativement à celle des particules légères, en faisant ainsi varier l’hétérogénéité. Cette simulation montre en particulier que le régime stationnaire temporel atteint en conditions hétérogènes ne correspond pas à une distribution spatiale homogène des radicaux.
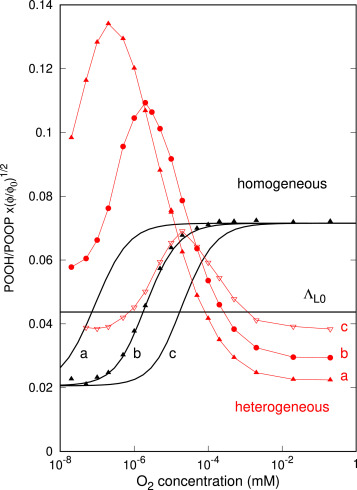
Le processus de radio-oxydation est intrinsèquement hétérogène au niveau sub-micrométrique, ce que néglige le modèle classique. La variation du rapport normalisé POOH/POOP avec la concentration en O2, contrôlée par la pression ambiante, montre une forte différence entre le modèle hétérogène (en rouge sur la figure) et le modèle idéalement homogène (en noir). Ce rapport simulé est en principe mesurable expérimentalement, ce que nous ferons par la suite, en faisant varier le TEL (Transfert d’Energie Linéique) des particules (et donc l’hétérogénéité), la pression et le flux. Par ailleurs, la méthode est évolutive et permettra d’intégrer des chaînes cinétiques plus complexes.
Contacts : Benoit Gervais et Yvette Ngono (CIMAP).
 |
La structure des défauts paramagnétiques de type P3 dans des verres phosphates révélée par calculs ab initioNadège Ollier (LSI) |
Les verres de phosphate ont plusieurs atouts, comme leur faible température de transition vitreuse Tg, qui leur confèrent de multiples applications allant de l’optique (matériaux lasers) à la médecine (verres bioactifs). Même si la structure des verres de phosphate a été largement étudiée, la nature exacte de certains défauts ponctuels générés sous irradiation comme le centre P3 par exemple, fait encore débat. Dans le cadre d’un projet du PTC Matériaux avancés entre la DAM et la DRF, nous avons simulé par dynamique moléculaire classique et par « Reverse Monte Carlo » (RMC) le verre de P2O5 avec une précision jusqu’ici inégalée (cellules de 112 atomes), ainsi que des verres binaires plus complexes Na2O-P2O5 (cellules de 100 atomes). Dans ces verres modèles, des lacunes d’oxygène paramagnétiques ont été générées afin de calculer par DFT leurs paramètres RPE (Résonance Paramagnétique Électronique).
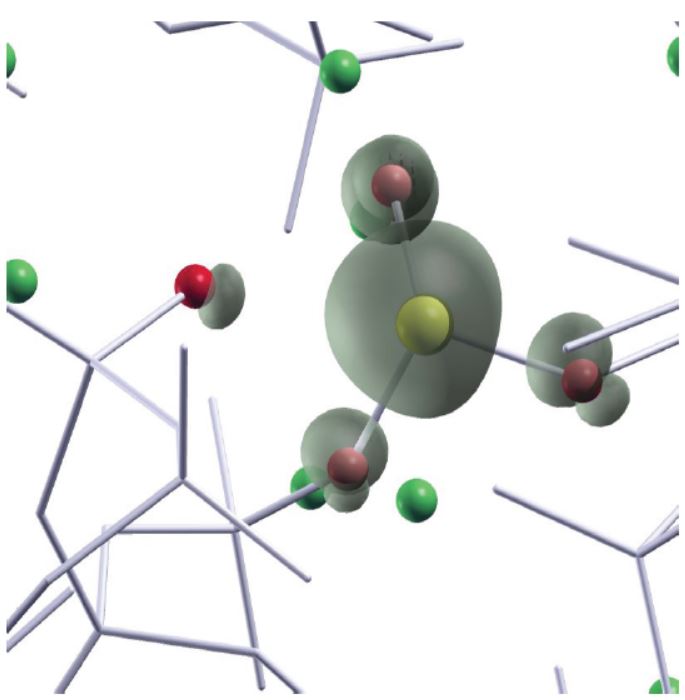
Ces calculs ab initio ont mis en évidence deux variantes de défauts P3 appelées P3a (un atome de phosphore lié à deux oxygènes pontants et un oxygène non pontant) et P3b (un atome de phosphore lié à un oxygène pontant et deux oxygènes non pontants), illustrées sur la figure. Nos calculs ab initio ont permis non seulement de résoudre la question de l’origine du défaut P3, mais d’expliquer également pourquoi le couplage hyperfin du défaut P3 obtenu expérimentalement variait avec la composition du verre (teneur en alcalin) ou la dose d’irradiation. En effet P3a et P3b ont des constantes de couplage hyperfin différentes, sensibles à l’environnement local.
Ce travail a été effectué dans le cadre d’une collaboration avec L. Giacomazzi (Materials Research Laboratory, Université de Nova Gorica, Slovénie et CNR-IOM Democritos Trieste, Italie).
Contact : Nadège Ollier (LSI).
