W. Chin, M. Mons
SPAM / Lab. Francis Perrin (URA CEA-CNRS 2453), CEA Saclay, 91191 Gif-sur-Yvette Cedex, France
Les protéines, molécules indispensables à la vie des organismes vivants, sont des polymères synthétisés à partir de l’ADN des cellules. Leur fonction dépend de leur constitution mais aussi de leur structure. Ces molécules sont constituées de longues chaînes linéaires d'acides aminés (typiquement une centaine) ; c’est ce qu’on appelle la structure primaire des protéines. Si l’on réalise que 20 types différents d’acides aminés existent dans le vivant et que chacun d’eux peut présenter plusieurs structures différentes, on conçoit que cette stratégie ait donné lieu à une très grande diversité de structures et de fonctions. L’un des points clés contrôlant la structure des protéines est le processus de repliement conduisant le polymère linéaire à adopter une structure tridimensionnelle. La première phase du repliement conduit à la formation de structures-type, appelées structures secondaires, concernant un nombre restreint d’acides aminés de 2 à quelques dizaines, dont les plus connues sont les feuillets β ou les hélices α. Le repliement se poursuit ensuite par l’agencement relatif des structures secondaires entre elles.
Comprendre et décrire le vivant, notamment avec les outils modernes de simulation numérique, nécessite donc d'appréhender ces organisations structurales et de connaître précisément les interactions physicochimiques qui les gouvernent. Une approche expérimentale nouvelle, développée dans l’équipe « Biomolécules excitées » du SPAM/LFP à Saclay, consiste à s’intéresser à de petits segments de protéines – les chaînes peptidiques- et à les faire se replier sous l’action de leurs propres interactions intramoléculaires. En pratique, on porte ces peptides en phase gazeuse à haute température dans un état désordonné « dénaturé », par une technique de désorption par laser, puis, dans une détente supersonique, on les laisse se refroidir par collisions avec le gaz porteur. Le repliement obtenu est alors analysé a posteriori, avec toute la précision des spectroscopies laser les plus modernes (technique de double-résonance IR/UV). Le croisement des informations spectroscopiques électroniques et vibrationnelles permet alors de caractériser les minimas locaux du paysage conformationnel balayé, notamment au travers de leur réseau de liaisons hydrogène intramoléculaires.
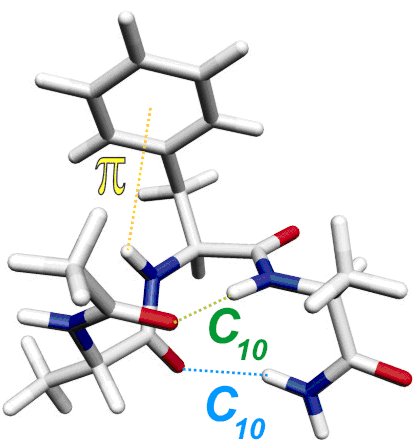
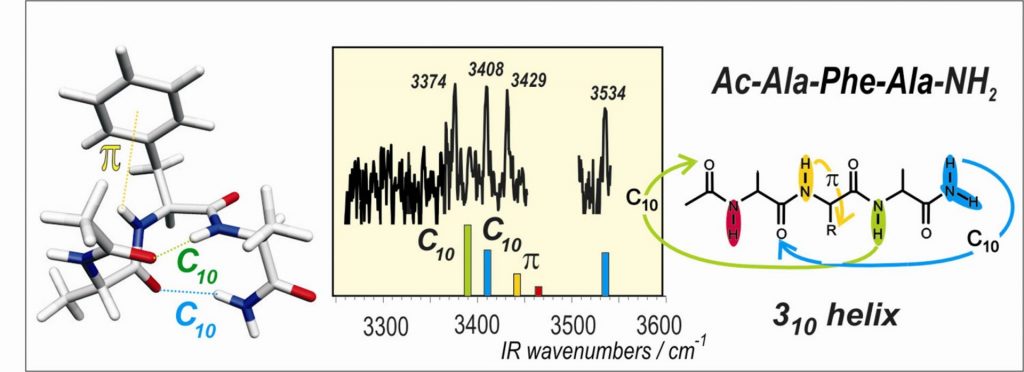
Ainsi plusieurs structures secondaires classiques des protéines ont pu être isolées dans la détente montrant bien le caractère intrinsèque de leur repliement, y compris en dehors de tout environnement biologique. Par ailleurs, ces structures ont été caractérisées avec une précision bien meilleure qu’en phase condensée, permettant de mettre à jour les interactions subies par chaque groupement NH dans chacun des conformères observés: une échelle de force des différentes interactions présentes dans ces systèmes a pu être établie.
Les coudes β responsables du repli des chaînes des protéines sur elles-mêmes, sont déjà observés dans les petites chaînes à deux acides aminés malgré la compétition avec les formes dépliées. Ils sont caractérisés par une liaison H relativement faible et doivent leur stabilité relative à un faible niveau de contrainte du squelette.
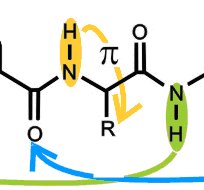
Les hélices 310 se forment spontanément dans les petites chaînes comportant trois acides aminés : Elles sont cependant très flexibles et sont en compétition avec des formes voisines. Cette compétition peut être notablement influencée par des interactions mineures entre les chaînes latérales et le squelette, comme les interactions NH-aromatique.
Une telle approche est complémentaire de la simulation par dynamique moléculaire car elle permet de valider celle-ci et plus précisément les potentiels intermoléculaires sous-jacents. Les résultats obtenus en phase gazeuse sont en effet les seules données expérimentales directement comparables aux meilleurs calculs de chimie quantique qui servent de référence pour le paramétrage de ces potentiels. L’observation de la transition entre hélice 310 et hélice β lorsque la chaîne s’allonge est le prolongement naturel de ces études, compte tenu de l’importance de cette dernière dans les protéines.
Au contraire des informations structurales recueillies en phase condensée cristalline, notamment par les techniques de diffraction de rayons X, les techniques de jet moléculaire et de spectroscopie optique, usuellement utilisées en physique ou en chimie, se révèlent extrêmement puissantes pour obtenir des informations locales sur les forces gouvernant la structure secondaire des chaînes de protéines. A ce titre elles permettent d’affiner les modèles décrivant les protéines et participent ainsi à l’effort interdisciplinaire de modélisation du vivant.
Références :
[1] W. Chin, J.-P. Dognon, F. Piuzzi, B. Tardivel, I. Dimicoli, and M. Mons,
J. Am. Chem. Soc. 127, 707 (2005)
[2] W. Chin, I. Compagnon, J.-P. Dognon, C. Canuel, F. Piuzzi, I. Dimicoli, G. Von Helden, G. Meijer and M. Mons,
J. Am. Chem. Soc. 127, 1388 (2005)
[3] W. Chin, F. Piuzzi, J.-P. Dognon, I. Dimicoli, B. Tardivel and M. Mons,
J. Am. Chem. Soc. 127, 11900 (2005).