G. Biroli*, J.-P. Bouchaud, D. L’Hôte, F. Ladieu (DSM/DRECAM/SPEC et *DSM/SPhT)
Le problème de la transition vitreuse est selon le prix Nobel de Physique P.W. Anderson « le problème le plus profond et le plus intéressant en physique de l’état condensé ». A première vue pourtant, il est difficile de penser que des matériaux aussi courants que les verres puissent encore constituer un « mystère » de la physique. Il n’empêche que la compréhension quantitative des verres et plus particulièrement de la transition vitreuse défie les physiciens depuis plus de cinquante ans.
Une équipe du SPEC montre l’importance d’une longueur de corrélation dynamique, qui caractérise le nombre de molécules liées dans leur mouvement de diffusion, et propose une méthode simple permettant de la mesurer à l’aide d’une généralisation des relations fluctuation-dissipation appliquées au problème.

Les verres représentent l’archétype de ce que l’on appelle aujourd’hui les « systèmes complexes ». Leur complexité est associée à un effet de « frustration » : l’existence de contraintes antagonistes empêche les particules (atomes, molécules) d’évoluer vers une configuration d’énergie minimale par une simple optimisation des structures locales. Cet effet est présent dans beaucoup d’autres situations, même en dehors de la physique. La théorie des systèmes vitreux se révèle donc fort utile pour décrire les verres physiques, mais aussi la matière granulaire, ainsi que des « verres » plus abstraits, qui apparaissent dans des disciplines à priori lointaines comme l’informatique, la théorie des jeux ou l’optimisation combinatoire.
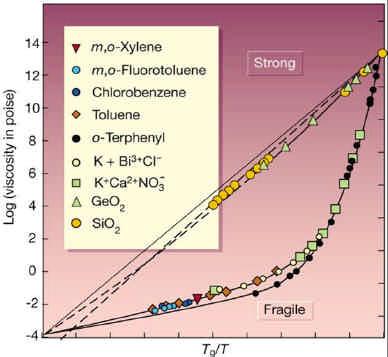
Un verre est obtenu à partir d’un liquide refroidi suffisamment vite pour qu’il ne cristallise pas et qu’il reste dans un état métastable appelé liquide surfondu. Si à partir de cet état on baisse encore la température, le temps de relaxation augmente extrêmement vite, jusqu’à dépasser les temps expérimentaux typiques en dessous de la température de transition vitreuse Tg. La Figure 1 montre une augmentation de treize ordres de grandeur pour une variation très modeste de la température au-dessus de Tg. Ce passage de l’état liquide au verre échappe aux théories qui ont pourtant décrit avec succès les transitions de phases de nombreux corps, comme la transition liquide-gaz ou paramagnétique – ferromagnétique. L’existence même d’un état vitreux bien défini et par conséquent d’une vraie transition vitreuse thermodynamique est toujours en débat ; il pourrait plutôt s’agir d’une transition entre deux régimes dynamiques différents.
Les scénarios envisagés pour la transition vitreuse, ainsi que les approches analytiques, numériques et expérimentales se sont multipliés ces dernières années. Une idée centrale est que l’évolution du système n’est permise que si dans certaines régions de l’espace, les particules « coopèrent » pour réorganiser leur structure. Cette idée permet d’expliquer l’augmentation brutale du temps de relaxation pour les liquides dits « fragiles » (Fig. 1) lorsque la température tend vers Tg. D’un point de vue expérimental, il est de plus en plus clair que la dynamique à l’approche de la phase vitreuse est très hétérogène : des régions lentes coexistent avec des régions rapides. Ce caractère hétérogène est associé à la nécessité de réorganiser collectivement des régions entières. Le ralentissement est alors associé à la croissance d’une longueur de corrélation dynamique, observée de façon précise dans des simulations numériques, mais seulement de manière très indirecte expérimentalement.
L’accès direct à cette longueur de corrélation dynamique dans les verres en formation en fonction de la température ou de la densité est donc un enjeu majeur. Nous avons proposé et mis en œuvre expérimentalement une méthode simple permettant de la mesurer : en utilisant une généralisation des relations fluctuation-dissipation (qui remontent, dans leur forme la plus simple, à l’article d’Einstein de 1905 sur le mouvement Brownien) aux fonctions de corrélation dynamiques. On montre que la dépendance en température ou en densité d’observables facilement accessibles, comme la constante diélectrique ou le facteur de structure dynamique, permet d’estimer la taille typique des hétérogénéités dynamiques : cette dépendance donne une mesure du nombre de molécules corrélées, qui peut être relié à la longueur de corrélation dynamique. La mise en œuvre de ces idées – sur le glycérol au CEA/SPEC, et sur un système colloïdal au Laboratoire des Verres de Montpellier – ont permis d’obtenir la première confirmation expérimentale directe qu’une longueur de corrélation dynamique croît bien à l’approche de la transition vitreuse, et d’estimer cette longueur quantitativement. Un exemple de résultat expérimental est reproduit dans la figure ci-contre : chaque courbe correspond, pour le glycérol (Tg = 185 K), à une mesure du nombre de molécules corrélées à une température donnée T. qui est bien une fonction croissante de |T-Tg|.
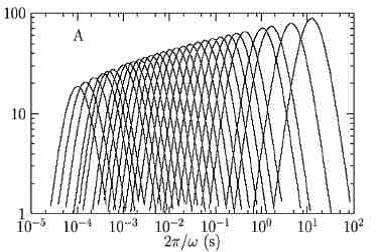
Fig. 2. Glycérol : Nombre de molécules corrélées au temps t pour passer d’une configuration microscopique à une autre. Chaque courbe correspond à la mesure à une température entre 232 K (à gauche) et 192 K (à droite). Le réseau de courbe montre l’augmentation de la longueur de corrélation dynamique à l’approche de Tg = 185 K.
Notre analyse, peut être appliquée à d’autres résultats publiés dans la littérature afin d’extraire la longueur dynamique pour un très grand éventail de matériaux à Tg. De manière assez inattendue, on observe que cette longueur est proche de six unités moléculaires, valeur pratiquement indépendante du matériau considéré. Ceci signifie qu’au voisinage de Tg, environ deux cents molécules doivent se coordonner pour qu’un verre puisse couler.
L’extension de notre méthode à une plus grande gamme de fréquence, à d’autres matériaux (comme les verres de spin) et en dessous de la température de transition vitreuse, devrait permettre de départager les nombreux scénarios théoriques proposés au cours des quarante dernières années pour décrire la dynamique anormalement lente des verres, et de soulever un coin du voile qui recouvre l’un des grands « mystères » de la physique statistique.
Référence :
Direct experimental evidence of a growing lengthscale escorting the glass transition,
L. Berthier, G. Biroli, J.-P. Bouchaud, L. Cipelletti, D. El Masri, D. L’Hôte, F. Ladieu, M. Pierno, Science, 310 (2005) 1797.
Contact : DRECAM/SPEC : F. Ladieu (Equipe GIT).