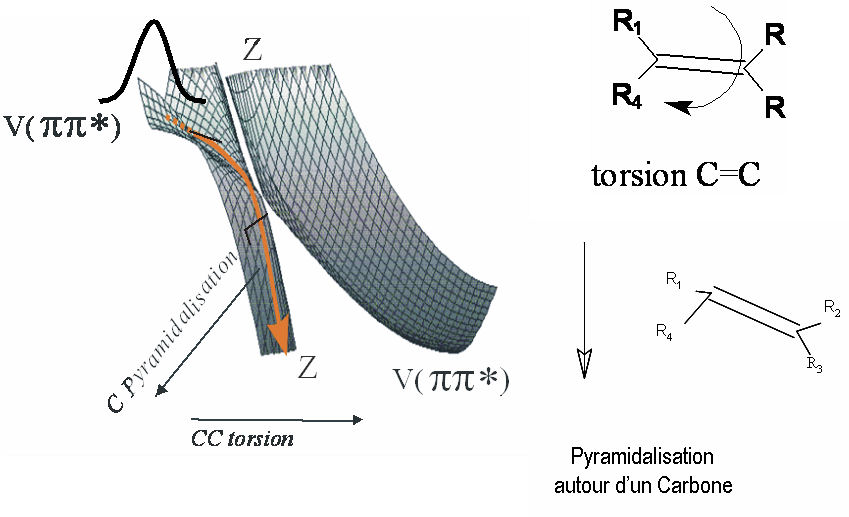
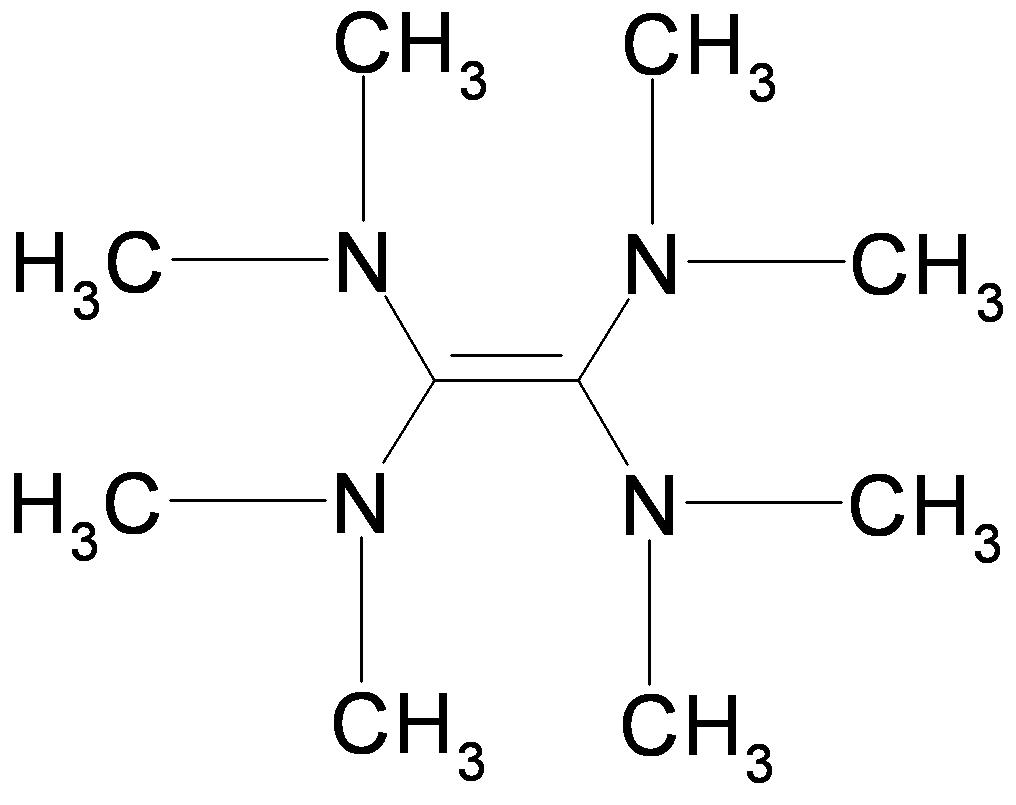
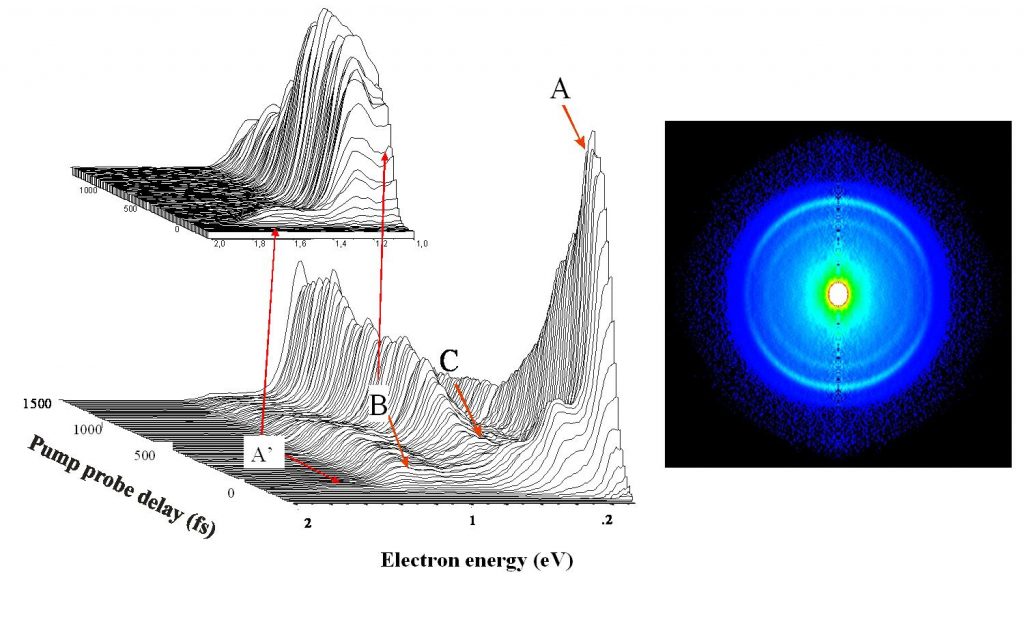
E. Gloaguen 1Laboratoire Francis Perrin (CNRS-URA-2453), DSM/DRECAM/ Service des Photons, Atomes et Molécules, C.E.A. Saclay, F-91191 Gif-sur-Yvette Cedex, France L’étape clef d'une réaction chimique est le passage du système réactionnel par un état transitoire, zone de non retour sur le chemin réactif qui relie les composés initiaux aux produits de réaction. Quand la réaction est photo – induite, la relaxation électronique qui précède l'arrivée à l'état de transition est une autre étape parfois cruciale pour déterminer le devenir de la réaction. En effet, il est rare qu'une molécule organique de grande taille, excitée électroniquement, réémette de la lumière ou réagisse à partir de son état excité initial. Des expériences de type pompe-sonde utilisant des lasers femtoseconde (10-15 s) ont montré que cette étape de relaxation est nettement plus rapide (inférieure à la picoseconde 10-12 s) qu'on ne l'imaginait. Ceci implique un changement de configuration électronique à l'échelle de temps des mouvements des coeurs atomiques[1]. Le mécanisme en jeu fait appel aux intersections coniques entre surfaces de potentiel. Ces points singuliers en forme d'entonnoir permettent la communication directe entre ces surfaces. Leur sont associés, des gradients de potentiel importants et des changements brutaux de configuration électronique. Au voisinage de ces points, des forces importantes sont appliquées à la molécule excitée qui rapidement se déforme et change de configuration électronique. Ce processus, propre aux systèmes polyatomiques, est essentiellement multidimensionnel. Il est illustré Figure 1 pour un alcène dont la configuration électronique change de V(ππ*) à Z (configuration C+-C–) pendant que la molécule se déforme selon deux coordonnées (torsion et pyramidalisation). Soulignons que les phénomènes de relaxation électronique à l’échelle femtoseconde et les passages par des intersections coniques qui en sont responsables offrent un cadre conceptuel couramment invoqué pour expliquer la non destruction photochimique des molécules biologiques quand celles-ci sont exposées au rayonnement solaire. Le groupe de dynamique réactionnelle du laboratoire Francis Perrin utilise une molécule modèle, le tetrakis (diméthylamino)éthylène (TDMAE) pour caractériser ces processus de relaxation de façon aussi directe que possible. En première approximation, la relaxation électronique de TDMAE excité à 4.6 eV dans l’état V(ππ*) se passe comme indiqué plus haut pour un alcène, via une intersection conique, vers un état Z, ce omportement étant une conséquence directe de la présence d'une double liaison C=C au centre de la molécule TDMAE. Les expériences réalisées utilisent l’imagerie de photoélectrons[2]. Ce nouveau diagnostic permet de connaître l'énergie et la distribution angulaire des électrons résultant de l'ionisation de la molécule par les photons sonde et apporte donc une signature des états électroniques par lesquels passe la molécule au cours de son évolution. Le résultat obtenu est très surprenant. Alors que, comme pour l'alcène, deux états électroniques V et Z étaient attendus, les résultats font clairement apparaître une autre étape (voir Figure 2). La bande A attribuée au déclin de l'état de valence V(ππ*) est accompagnée de la montée de la bande B, laquelle décline en même temps que monte la bande C attribuée à l'état Z. L'intermédiaire B, qui apparaît clairement ici, peut être attribué à un état de Rydberg qui joue le rôle d’un adaptateur de forme entre l’orbitale initiale et l’orbitale finale impliquées dans le changement d’état électronique V(ππ*) vers Z. Ainsi le schéma simple impliquant une relaxation directe entre les configurations V et Z doit-il être complété par le rôle joué par les états de Rydberg énergétiquement proches de l'état de valence. Ce nouvel effet pourrait avoir une certaine généralité car de nombreuses molécules possèdent un ou plusieurs états de Rydberg proches et facilement couplés à l'état de valence. On peut penser que le rôle de l'état de Rydberg est d'offrir à l'électron excité une orbitale d'accueil de forme intermédiaire aux orbitales impliquées dans les états V et Z de la molécule. Reference: [1] « Prereactive evolution of monoalkenes in the 6eV region » M.Elhanine, J.M.Mestdagh ,J.P.Visticot B.Soep J.Chem.Phys.113,237 (2000) [2] « Experimental Evidence for Ultrafast Electronic Relaxation in Molecules, Mediated by Diffuse States » Gloaguen E., Mestdagh J.-M., Poisson L., Lepetit F., Visticot J.-P., Soep B. J. Am. Chem. Soc. 127, 16529, (2005)
2 Department of Molecular and Laser Physics, University of Nijmegen Toernooiveld 1, 6525ED Nijmegen, Pays-bas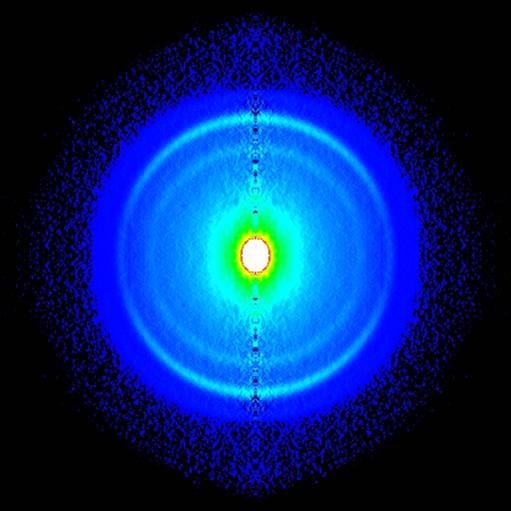
![Participation d’un état de Rydberg à la redistribution ultrarapide de l’énergie électronique du TDMAE [tetrakis(diméthylamino)éthylene]](https://iramis.cea.fr/wp-content/uploads/2004/10/972_1-2-300x300.jpg)