
invited by Yannick DAPPE
Abstract :
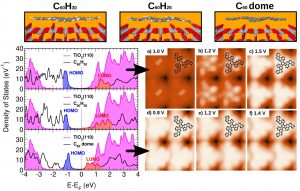
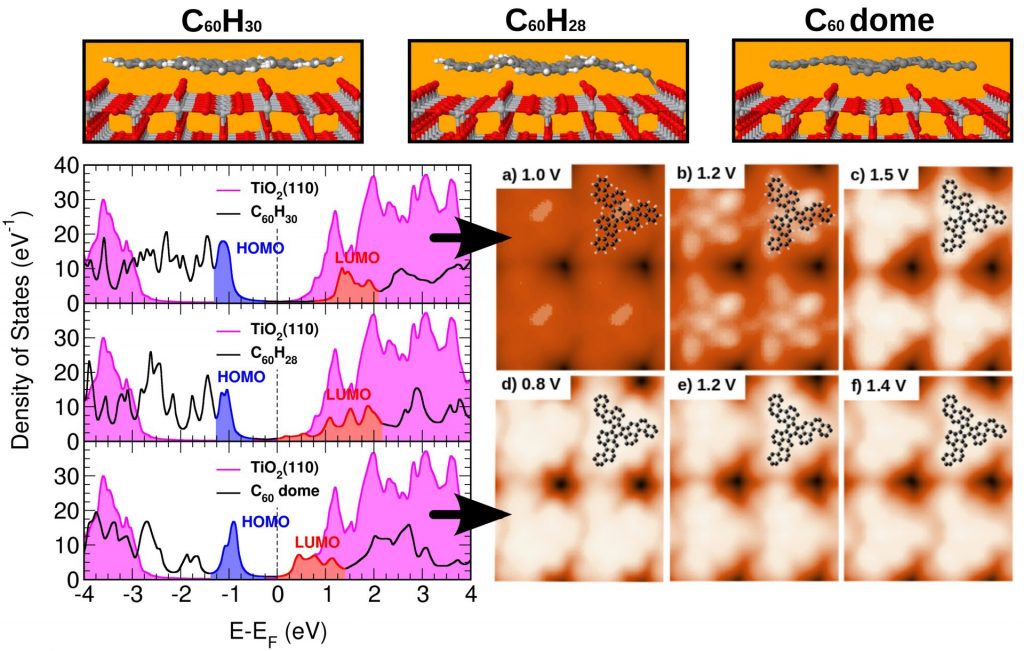
Understanding the interaction between organic molecules and metal surfaces is of paramount importance in diverse fields such as organic electronics, molecular electronics, catalysis, surface photochemistry, among others. In particular, the growing field of organic electronics relies on the use of organic conjugated molecules as components of multilayer devices. The performance of these devices depends critically on the energy barriers that control the carrier transport between layers, energy barriers that are determined by the relative alignment of the molecular levels at substrate–organic (MO) or organic–organic (OO) interfaces. It has been suggested that the tendency of the Charge Neutrality Level (CNL) of the organic material to align with the interface Fermi level plays a crucial role; this mechanism is associated with the induced density of interface states (IDIS) in the organic gap and the charge transfer between the organic and the substrate. More recently, this model has been extended to the Unified IDIS model by inclusion of Pauli repulsion and intrinsic molecular dipoles. On the basis of the Unified IDIS model, we have investigated the interaction of a variety of organic molecules such as Bz [1a], TTF [1b], TCNQ [1c], PTCDA [1d, 1e], and some Heteroaromatics (C60H24N2 and C60H30) [4], with essentially different substrates, such as Au(111), TiO2(110) and supported Graphene. These molecules are prototypical electron donors and acceptors and are in potentials of molecular electronics as promising candidates of organic conductors, and some of them interesting due to their hole injection barrier found on a variety of polycrystalline metal substrates. On the other hand, thermal-induced (cyclo)dehydrogenation leads some Heteroaromatic PAH precursors to the formation of pristine or N-doped graphene [4]. In our theoretical approach we first obtain the geometric configuration of the substrate / organic interface by using a powerful molecular dynamics DFT-based technique [1b], combined to an effective van der Waals correction in a perturbative fashion. The electronic structure and substrate / organic barrier height formation for the single molecules, as well as for the ad-layered structures, are analyzed by using the mentioned IDIS model [1b, 2], applying to the whole system a convenient energy level alignment. Within this framework we calculate the charging energy Ueff of all the isolated and ad-layered systems at the nanocontacts, determining self-consistently the transport energy gap and the organic molecule density of states. An accurate Keldish-Green-based theoretical scanning tunneling microscopy approach [2] has been performed over all the obtained structures in order to compare the results with those existing in previous literature and recent experiments by our group, finding an excellent agreement between experiments and theory [1a-e, 3a, 3b, 4].
References
1. a) J. I. Martínez et al., J. Chem. Phys. 134, 044701 (2011); b) Org. Elec. 13, 399 (2012); c) Phys. Stat. Sol. B 248, 2044 (2011); d) Chem. Phys. 390, 14 (2011); e) Phys. Rev. Lett. 108, 246102 (2012).
2. J. M. Blanco et al., Phys. Rev. B 70, 085405 (2004).
3. a) I. Fernández-Torrente et al., Phys. Rev. Lett. 99, 176103 (2007); b) Int. J. Mass Spectr. 277, 269 (2008).
4. A. L. Pinardi, J. I. Martínez, J. A. Martín-Gago et al., ACS Nano, DOI: 10.1021/nn400690e (2013).
Dept. de Superficies y Recubrimientos (ICMM / CSIC), ES-28049 Madrid, Spain