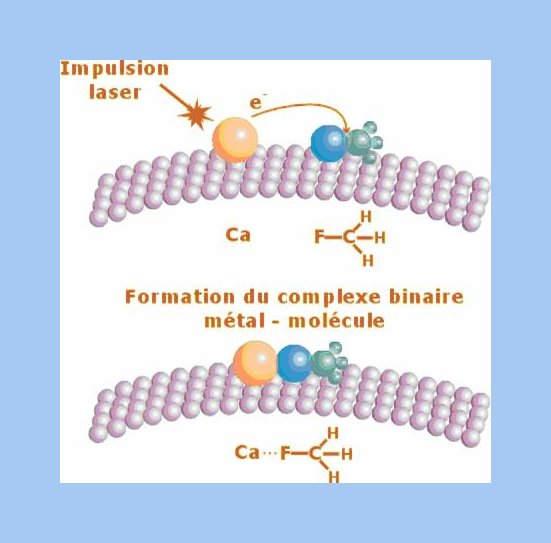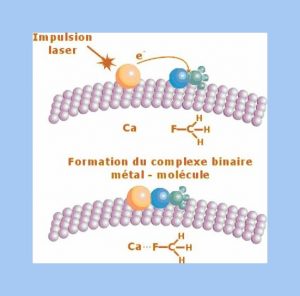
A chemical reaction depends not only of atoms and molecules involved but also of their short range environment. Understanding a chemical reaction demands a fundamental approach taking into account both temporal and spatial features.
Therefore, IRAMIS implements with lasers, time-resolved spectroscopies in the range from femtosecond to the millisecond, to study the dynamics of molecular systems, like for example DNA biomolecules, or chromophore molecules for photovoltaics. The various conformations adopted by the biomolecules by virtue of their flexibility are studied by a dual experience – theory approach, with quantum chemical simulations. More complex systems that are out of equilibrium, including isolated gas phase or molecular systems bound to aggregates, are also studied in order to identify and model the forces that drive their reaction dynamics.