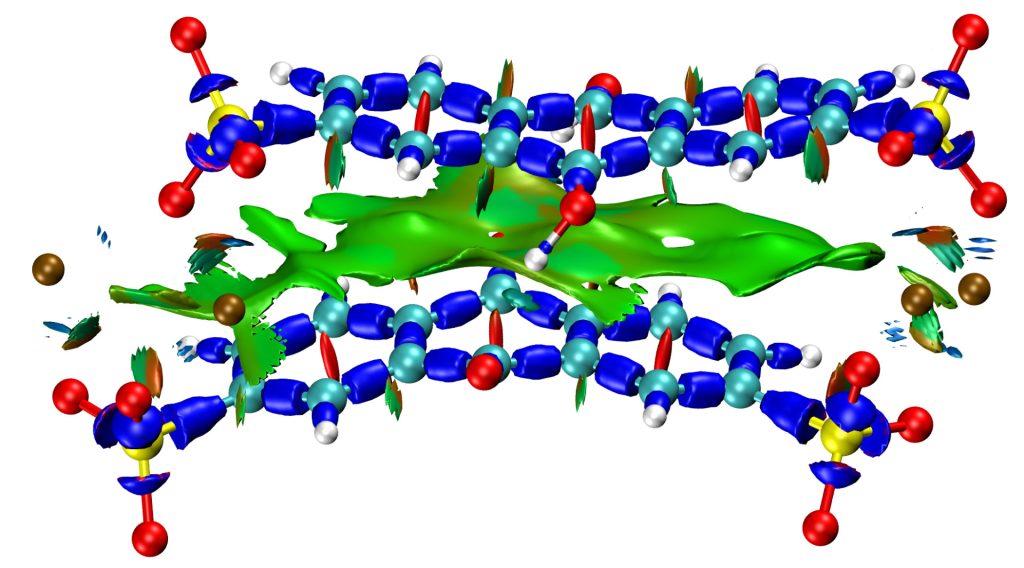
Simulation for solid-state NMR
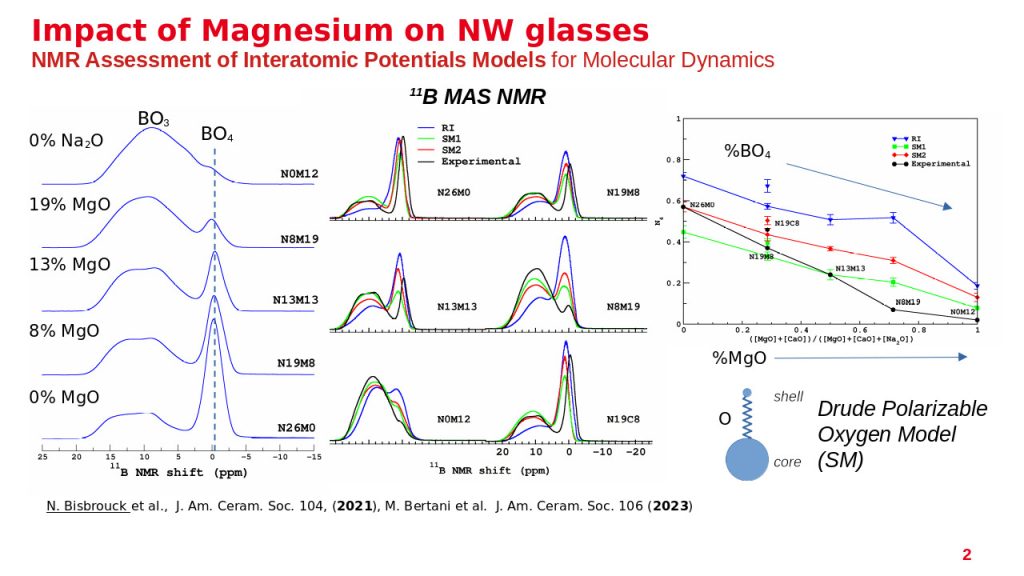
In order to support our experimental NMR investigations of glasses (and other materials in general), we develop computational methodologies based on the combination of molecular dynamics simulations with DFT computations of NMR interactions for improving our understanding of collected NMR spectra. These numerical methodologies allow us to predict not only NMR experiments (MAS NMR and MQMAs spectra) and to assess structural models, but also the NMR fingerprints of individual local environments of the atom of interest. Ongoing developpement focus on the acceleration of these DFT computations with machine learning, and their integration in reverse Monte Carlo approaches for modelling structures compliant with the NMR data.
Contact: Thibault Charpentier
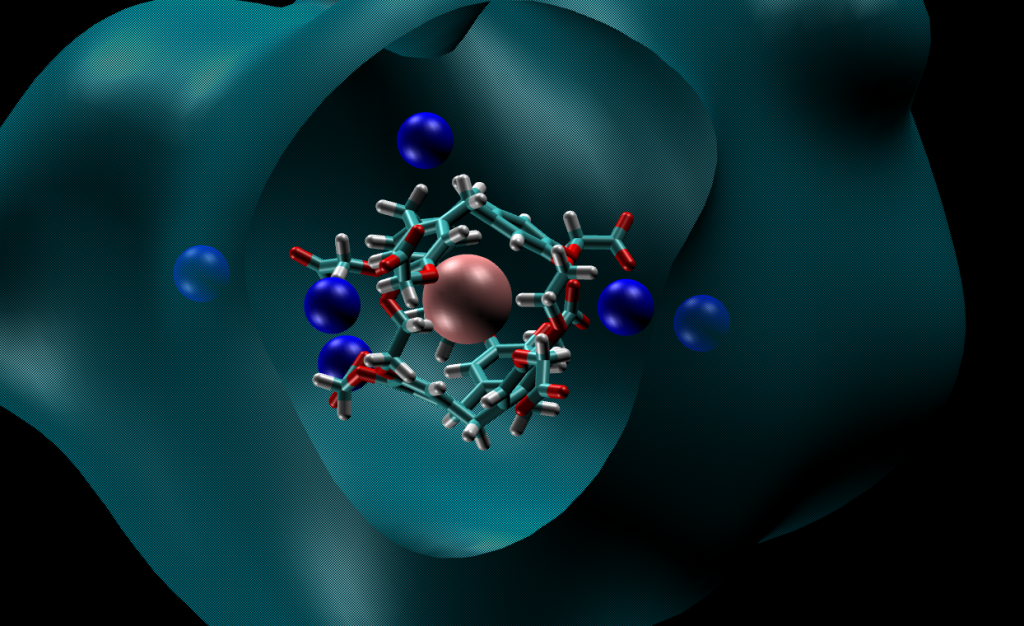
Simulation of liquid solutions
The subtle interactions between solute and solvent molecules or the breaking and formation of covalent bonds require the combination of electronic structure calculation and molecular dynamics under given thermodynamic conditions. To reduce the computational cost of these ab initio molecular dynamics (AIMD) simulations, we can use machine-learned potentials trained on ab initio energies and forces. In support to experiments, we predict structures, reaction mechanisms, or dynamical properties. Examples of applications at LSDRM concern hyperpolarized 129Xe in cryptophanes, nuclear glass alteration by water, or water-in-salts electrolytes for lithium-ion batteries.
Contact: Rodolphe Pollet
Molecular quantum chemistry
Although the numerical approach is mainly oriented towards NMR (design of innovative biosensors), several other themes describe the theoretical activity of LSDRM in the field of molecular chemistry, some of which involve methodological developments in the laboratory. They have the common characteristics of providing complementarity to the experiments carried out while initiating new original approaches. The applications are in the field of optoelectronics, battery aging, calculation of magnetic properties and in a more fundamental context, the definition of new chemical species. They implement a set of theoretical methods for exploring conformational space, for studying the electronic structure (mono or multireference wave function, 4-component relativistics) and topological methods for analyzing electron density.
Contact: Jean-Pierre Dognon