
In photovoltaic cells, the absorption of a photon by a semiconductor material creates an electron-hole pair (called an exciton), resulting from the excitation of an electron from the valence band to the conduction band. If the energy of the absorbed photon is greater than that of the semiconductor’s band gap, the electron-hole pair formed possesses excess energy that is rapidly dissipated, typically in the form of heat (relaxation at band edges). Extracting “hot” charge carriers (i.e. before they relax) could double the efficiency of photovoltaic devices.
In luminous devices such as LEDs or lasers, rapid relaxation of hot charge carriers is more important. It is therefore important to understand the mechanisms of electronic relaxation.
For all these applications, halogenated perovskite materials offer particularly interesting optical and electronic properties. These properties have also recently been studied in the case of nanostructures, where confinement effects radically alter the electronic structure of the material, which must influence relaxation rates. In the DICO team at LIDYL, the role of quantum confinement on relaxation has been explored by means of a femtosecond optical spectroscopy experiment (1 fs = 10-15 s): in perovskite nanoplatelets a few monolayers thick, it has been shown that the relaxation time remains very short (a few hundred fs). While nanostructuring-induced electronic level spacing should mitigate the main source of dissipation via phonon coupling, these results highlight that another particularly efficient relaxation pathway exists in strong confinement, and appears to be linked to the ligands on the surface of these colloidal nanostructures.
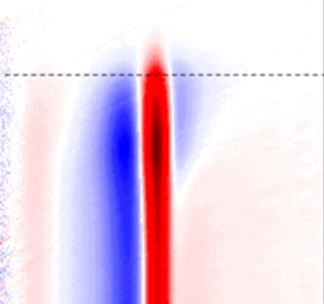
Halogenated perovskites, of the general formula ABX3, are attracting a great deal of attention for their optoelectronic properties, particularly in their halogenated form, where A is a cation, B a divalent metal, and X- a halide anion (I-, Br- or Cl-). Optical excitation with energy above the gap in these polar semiconductors leads to the formation of an electron-hole pair with excess energy. In this case, relaxation then takes place, typically in the form of a de-excitation cascade, by successive phonon emission, until total dissipation of the excess energy. The relaxation time depends not only on this excess energy, but also on the excitation density.
As part of the ANR “CaMPUUS” Accueil de Chercheurs de Haut Niveau (ACHN) project (in collaboration with LIDYL’s ATTO group), the sub-picosecond relaxation of electron-hole pairs in colloidal perovskite nanostructures has been studied on the Sofockle laser platform using a transient absorption technique. This consists in measuring the absorption difference ΔA(λ,t) of the sample, measured from the transmitted spectrum of a broadband laser pulse, preceded or not by a pump pulse, and as a function of the variable delay t between the 2 pulses. The spectra are recorded at a rate of 3 kHz and the associated dynamics measured with a time resolution of around 90 fs.
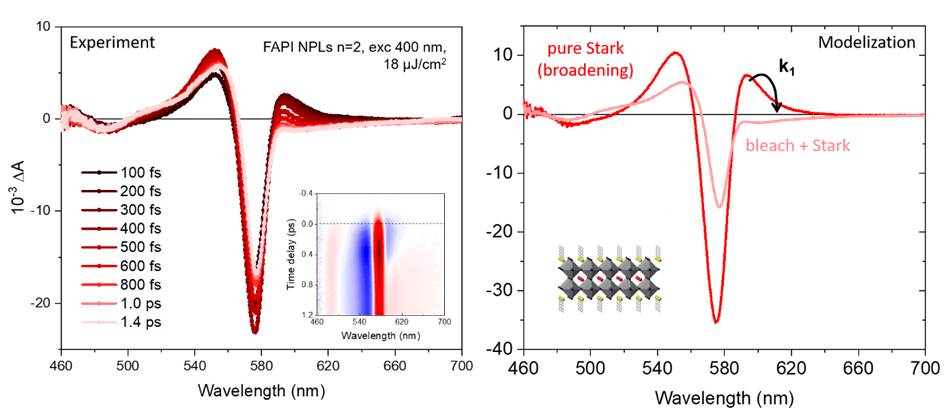
Left: experimental data (after corrections), with complete 2D t- λ map inset. Right: modeling of the relaxation from the “hot” state (red curve), showing a broadening of the absorption peak due to the Stark effect, to the final relaxed state (pink curve), essentially showing an attenuation of the absorption. The characteristic time is 1/k1 = 270 fs.
For this study, we first developed syntheses of colloidal perovskite nanostructures dispersed in solution: FAPbI3 (FA= formamidinium) nanocrystals with low quantum confinement (size approx. 11 nm), as well as two-dimensional nanoplatelets of variable composition and thickness defined to the nearest atomic monolayer (0.6 to 1.8 nm). Transient absorption measurements are then analyzed using a global spectral and kinetic fitting method (see figure) to extract relaxation times. The high Stark effect and the discretization of electronic levels in these confined systems prevent the use of conventional analysis methods, such as extracting charge carrier temperatures.
The reduction in size of objects down to the nanometric scale results in a spacing of electronic levels that can become far greater than the typical energy of vibrational modes. This energetic mismatch with phonons should then lead to longer relaxation times (“phonon bottleneck” effect), making perfectly resonant multiphonon emission unlikely. However, the measured time constants show that relaxation remains ultra-fast, becoming even shorter for the most confined nanostructures. This result can be explained by the increasing coupling of the exciton (electron-hole pair) with the vibrational modes of the ammonium ligands present on the nanostructure surface. This coupling becomes even stronger as the exciton wave function is delocalized outside the crystal structure.
In perspective, this work shows the importance of better controlling surface effects, to master the relaxation of hot carriers, and thus improve photovoltaic devices, where long relaxation times are sought, or light-emitting devices (LEDs or lasers), where fast relaxation times are sought.
This work is part of Carolina Villamil Franco’s PhD thesis (ED 2MIB 2017-2020).
Contact ERL LIDYL/DICO CEA-CNRS: Elsa Cassette.
Collaboration :
- Benoît Mahler, Institut Lumière Matière (ILM), UMR 5306 Université Claude Bernard Lyon I, CNRS, 10 rue Ada Byron, 69622 Villeurbanne Cedex, France
- Gaëlle Allard, Laboratoire Lumière-Matière aux Interfaces (LUMIN), FRE2036 Université Paris-Saclay, CNRS, ENS Paris-Saclay, France.