Ionized molecules are involved in many chemical reactions, and participate for an important part in the chemistry of the upper atmosphere and interstellar clouds. Data on the vibrational spectroscopy of these ions are thus needed to better understand the dynamics and energetics of so diluted matter.
Photoelectron spectroscopies are method of choice to characterize these molecules and their vibrational states, but are often ineffective when the structure of the neutral molecule is very different from that of the ion. The Laboratoire Francis Perrin (URA 2453, CEA – CNRS) in collaboration with the team of Chimie Théorique du Laboratoire Modélisation et Simulation Multi Echelle (MSME UMR 8208 CNRS, Univ Paris-Est Marne-La-Vallée) participated in the development of a new spectroscopic method to achieve the desired data, not accessible by conventional methods.

In the method, called SPES (Slow Photo-Electron Spectroscopy), the molecules are excited to an autoionizing state (Rydberg state) by a photon in the VUV (vacuum UV) range, and the non-zero energy electron with its associated ions are measured in coincidence after ionisation. The coincidence measurement allows selecting only the electrons from the ionization of Ar2. The non-zero energy of the electrons implies that the process of auto-ionization is associated with some vibrational relaxation, giving access to all the lower vibrational levels of the ionized molecule (Figure 1).
Thus, the originality of the method is based on the observation of the photoelectrons, after excitation of an auto-ionizing state followed by ionization. The vibration spectrum of the ion is obtained by a treatment of the photoelectron spectrum knowing the energy of each emitted electron.
The performance of this method have been highlighted by the study of argon dimers using the spectrometer coincidence imaging electron / ion installed on the beamline DESIRS of the synchrotron SOLEIL [1]. Atomic argon having all its electronic layers completed, Ar2 dimer can be stabilized only by van der Waals bonding, and its ground state has therefore a large interatomic distance. However, its Rydberg states and ion can lead to the formation of (metastable) excimer molecule or stable ionized molecule. So that the ground state and the ionized state are very different.
[1] DESIRS (Dichroïsme Et Spectroscopie par Interaction avec le Rayonnement Synchrotron) is a beamline based on an ondulator covering the VUV range (5-40 eV) with the required high resolution for detailed spectroscopic studies.
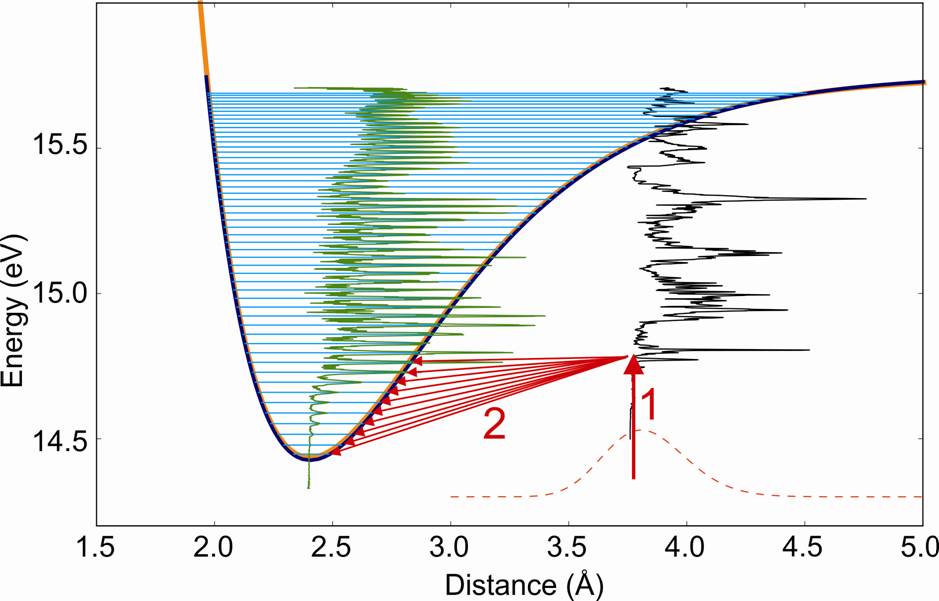
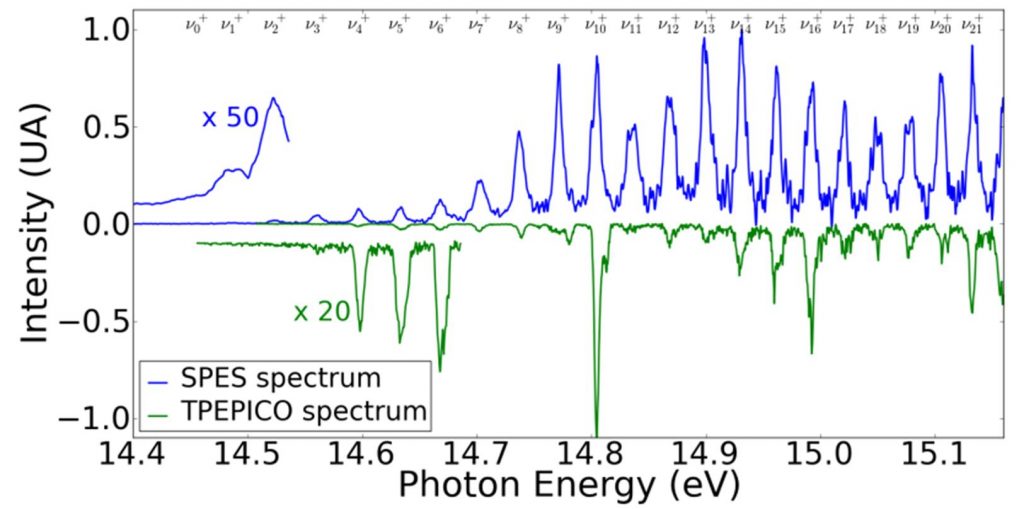
By applying the SPES method to this dimer, the lowest vibrational states of Ar2+ (see Fig.2) have been directly observed for the first time, while they are a priori unattainable according to the Franck-Condon principle (for which transitions are most effective between states whose vibrational wave functions show a strong spatial overlap). The potential energy curve of the Ar2+ ion, further obtained by reconstruction (RKR method, Rydberg-Klein-Rees), is in good agreement with that obtained by the best methods of current ab initio calculations. However, the small differences observed in vibrational spectroscopy, encourage further theoretical investigations. In addition, we also obtained the ionization probability for all vibrational levels accessible from each intermediate excited state.
The SPES method so validated on the model case of the Ar2 molecule is generic and can be applied to other systems, such as polyatomic molecules of atmospheric interest or biological, where electronic redistribution induced by ionization can lead to strong structural rearrangements.
References:
The SPES (Slow Photo-Electron Spectroscopy) method:
– Photoionization of 2-pyridone and 2-hydroxypyridine.
J.C. Poully, J.P. Schermann, N. Nieuwjaer, F. Lecomte, G. Grégoire, C. Desfrançois, G. A. Garcia, L. Nahon, D. Nandi, L. Poisson and M. Hochlaf, Phys Chem Chem Phys. 12 (2010) 3566.
– Ar2 photoelectron spectroscopy mediated by autoionizing states,
M. Briant, L. Poisson, M. Hochlaf, P. de Pujo, M.-A. Gaveau, and B. Soep, Phys. Rev. Lett. 109, (2012) 193401.
Contact: