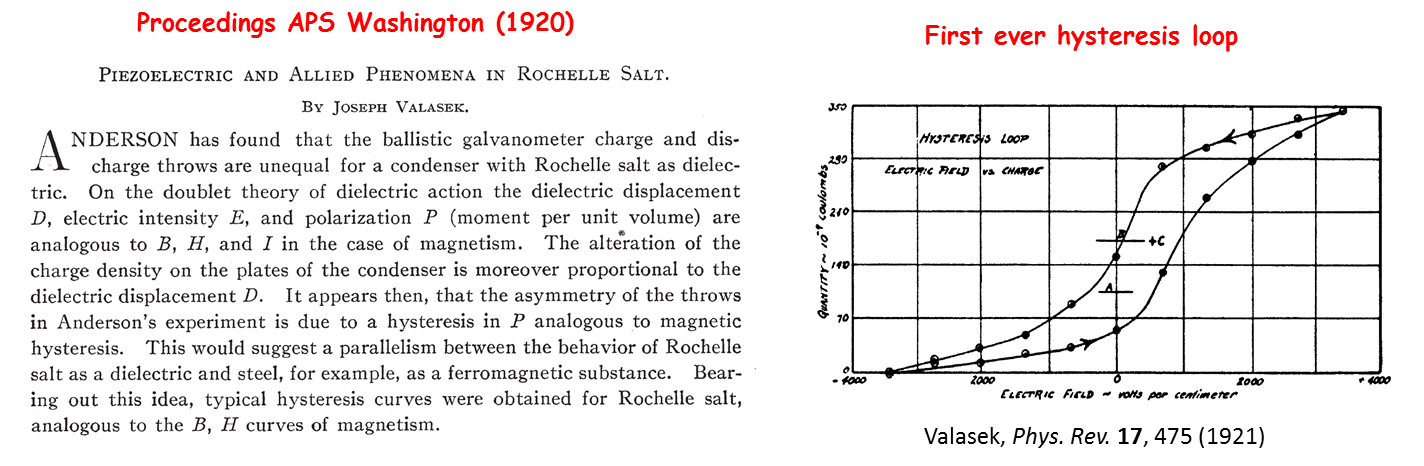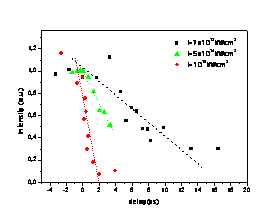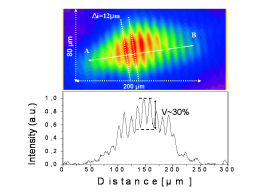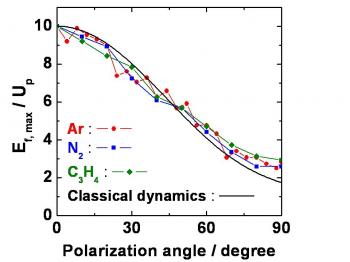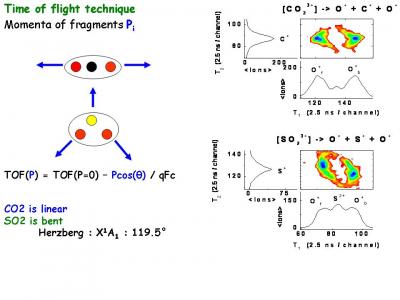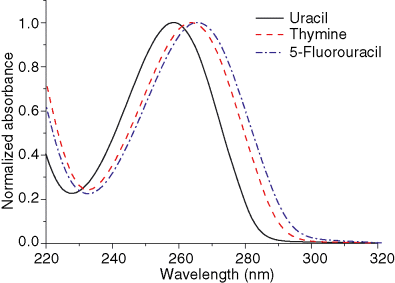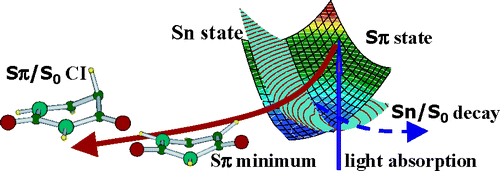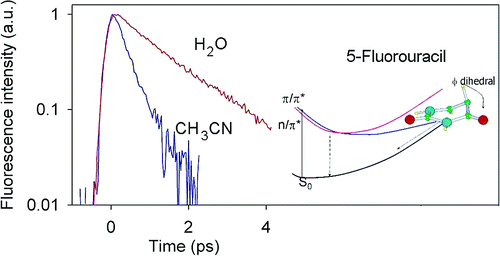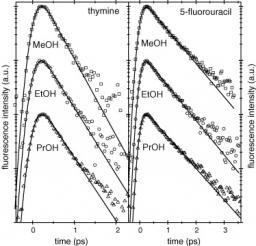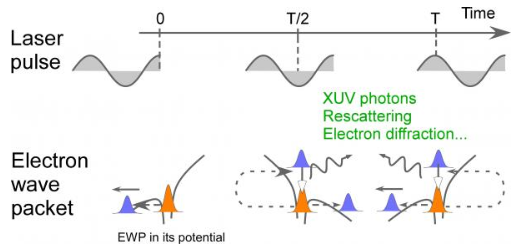
Atoms in a strong laser field: an electron wave packet is launched and driven by the field over one optical cycle. The EWP can return to its parent ion and be scattered as an outgoing electron wave or an attosecond burst of XUV light. The EWP recollision has therefore a double interest: it can be exploited either as a probe of the system with an extreme resolution, or as an ultra-short source of XUV light.
The dynamics of atomic and molecular electrons in a strong laser field is particularly rich and has been a central research topic at LIDYL for more than thirty years. This program is experimentally and theoretically continued in the Attophysics group.
Basically, the ultra-fast electron dynamics in a strong laser field can be described from both quantum and semi-classical concepts, as for instance electron wave packets and electron trajectories, respectively. The semi-classical picture has popularized the elementary dynamical process under the so-called “three-step” model [P. Corkum, Phys. Rev. Lett. 71, 1994 (1993)].
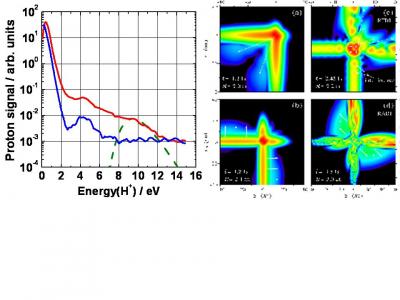
Its profound physical content is illustrated in the figure. When the atom (molecule) is submitted to a laser field – in the intensity range 1013-1016 W/cm2, that is strong enough to distort significantly the core potential -, an electron initially in a valence orbital can escape the core (step 1). This electron subsequently "rides" the laser field and may return back to its parent core within one optical cycle (of duration 2.7 fs = 2.7 10-15 s with the infra-red lasers we use), after it has gained kinetic energy in the field up to a few tens or even hundreds of electronvolts (step 2). In the recollision with the core, the electron can be quasi-elastically scattered (electron diffraction) or inelastically but coherently scattered (step 3). In the latter inelastic recollision, the electron can either further ionize the core or recombine radiatively with it, releasing its energy as an attosecond burst of extreme–UV light. The above three steps including the attosecond emission constitute the elementary sequence in High Harmonic Generation or HHG, first observed in 1987 simultaneously in Chicago and Saclay [A. Mc Pherson et al., J. Opt. Soc. Am. B 4, 595 (1987), M. Ferray et al., J. Phys. B 21, L31 (1988)]. Each optical cycle drives two recollisions so that a train of attosecond pulses in produced in HHG; their temporal characterisation was first achieved in Saclay in 2001 [P.-M. Paul et al., Science 292, 1689 (2001)].
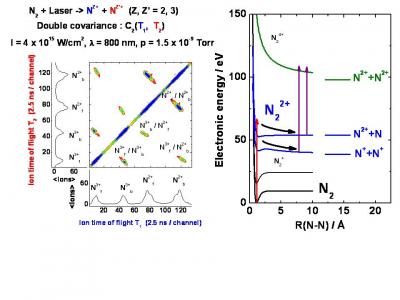
The atomic/molecular electron dynamics in the strong field encompass basic processes, such as ionization and EWP scattering in the different channels, which are studied for themselves along by two research lines. They are detailed in the Multiple ionization & Molecular Imaging and High Harmonic Generation and Attosecond physics pages.
Now, speaking quantum mechanics, the electron is better described as an electron wave packet (EWP) that dynamically splits into two parts, respectively bound and quasi-free, in the laser field, where the quasi-free component undergoes the recollision and scattering onto the core. The free EWP has a de Broglie wavelength in the Angstrom range, which makes it a very appropriate local probe of the system which extends over a comparable scale. Since the EWP probe has attosecond temporal resolution, it can in principle image ultra-fast motion of electrons and nuclei in molecules. Two research lines, described in the Ultra-fast imaging of molecules from electron diffraction and High Harmonic Generation and Attosecond physics pages, build on this "self-probing" paradigm.

Recolliding EWP in a strong laser field. The two coherent scattering channels, EWP diffraction and EWP radiative recombination keep an imprint of the nuclear structure and the electronic orbital in the molecule.
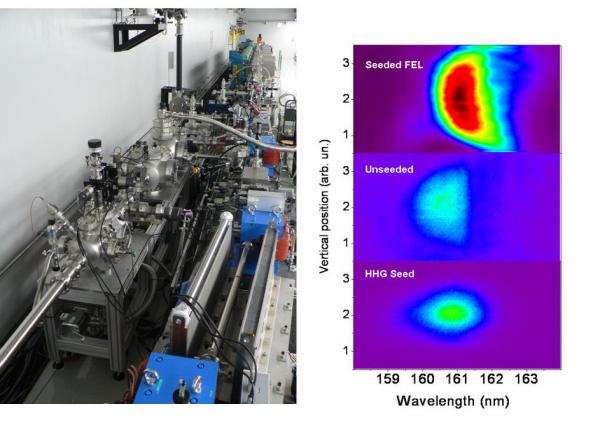
Besides the fundamental studies of the electron dynamics in strong field, and its use as a probe of transient systems, HHG provides with a source of ultra-short coherent pulses in the XUV (from 100 nm down to a few nm). The source's brightness, which reflects the high instantaneous flux and coherence in both the "narrowband" femtosecond and "broadband" attosecond ranges and its natural synchronization with a driving laser, make it very attractive for a number of applications. Among the Examples of applications we have performed multi-color Photoionization in the gas phase, and studies of XUV/solid interaction in the solid state. The coherence properties and partial tunability of the HHG source make it attractive for Seeding a Free Electron Laser, which constitutes another research line. A promising new application concerns the Coherent diffraction imaging of nanometric objects. Most of the applications are developed in collaboration with expert groups, either in France or in Europe, USA, Canada, Japan,…
Eventually, we pursue a theoretical activity to support the several experimental programs. It focuses on microscopic aspects of the gas phase-strong field interaction, i.e., the electron dynamics in atoms and molecules, including Strong Field Approximation (SFA) models in HHG. It also deals with the macroscopic aspects of the interaction, with the development of 3D propagation codes for the laser and XUV fields.