


This last decade has seen growing interest in the targeting of biological receptors by the encapsulation of xenon in functionalized host systems. The concept of smart xenon biosensors has been proposed, according to which the chemical shift of caged xenon varies when the host system is in the presence of the targeted receptor or analyte. A detailed understanding and reliable modeling of the interactions responsible for such effects on the resonance frequency of caged xenon are therefore of high value.
In this context, theoretical chemistry is useful to bring additional information to experiments and even by providing in certain cases experimentally inaccessible data.
Recently, for the first time, a thorough understanding of host–guest interactions, as well as their influence on the NMR spectroscopic parameters was gained in a 129Xe-cryptophane molecular system. All the components of the interaction between xenon and cryptophane were identified, and the origin of the measured 129Xe chemical shift value determined [1]. From a theoretical point of view, gaining an understanding of the nature of host-guest interactions in supramolecular complexes involving heavy atoms is a difficult task. First, due to the nature of xenon and the NMR properties to calculate, relativistic all-electron density functional theory (DFT) calculations are mandatory. Second, traditional density functionals do not include dispersion and are not fully appropriate for the description of the xenon-cage interaction. To recover most of the interaction, explicitly correlated methods are required.
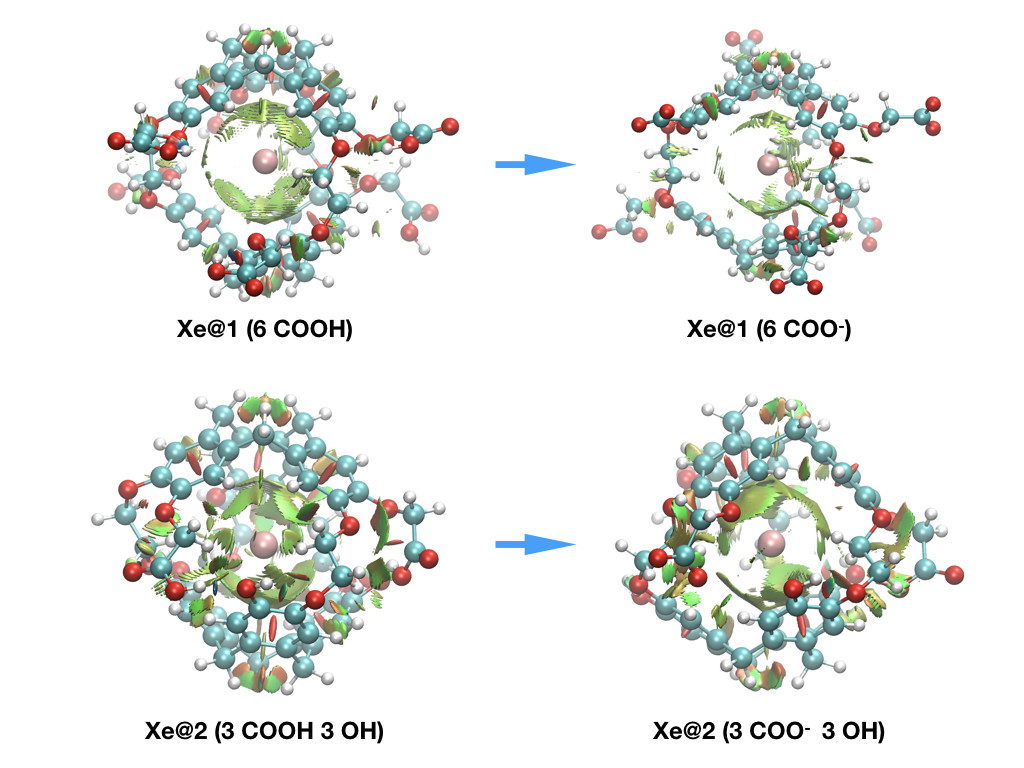
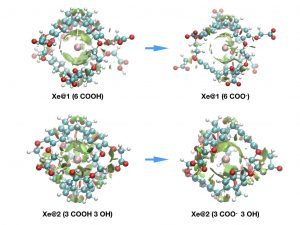
This approach, was also recently successfully applied to pH sensing using hyperpolarized 129Xe NMR Spectroscopy to provide explanation at the molecular scale for the origin of the chemical shift behavior [2].
To summarize, supplementing NMR chemical shift calculations, complementary-orbital, topological, and energy-decomposition analyses are powerful tools to understand host-guest interactions [3,4].
References
[1] Understanding a host–guest model system through 129Xe NMR spectroscopic experiments and theoretical studies
E. Dubost, J.-P. Dognon, B. Rousseau, G. Milanole, C. Dugave, Y. Boulard, E. Léonce, C. Boutin, P. Berthault, Angew. Chem. Int. Ed. 2014, 53, 9837-9840.
[2] Accurate pH sensing using hyperpolarized 129Xe NMR spectroscopy
E. Léonce, J.-P. Dognon, D. Pitrat, J.-C. Mulatier, T. Brotin, P. Berthault, Chem. Eur. J. 2018, 24, 6534-6537.
[3] Electronic structure theory to decipher the chemical bonding in actinide systems,
J.-P. Dognon, Coord. Chem. Rev. 2017, 344, 150-162.
[4] Theoretical insights into the chemical bonding in actinide complexes
J.-P. Dognon, Coord. Chem. Rev. 2014, 266–267, 110-122.