







2011 Année de la Chimie à l'IRAMIS
|
Le dossier "Chimie à l'IRAMIS " au format PDF
|
 |
|
Contact : Lionel Poisson (SPAM) |
- Relaxation ultrarapide : état médiateur et effet de solvatation
- Relaxation femtoseconde des métalloporphyrines
- Dynamique des molécules photochromes



Ruthénium Tétraphenyle Porphyrine Carbonyl (Ru TPP CO)
Dans le milieu vivant, les mécanismes, comme la respiration, producteurs d’énergie via le transport de petites molécules (O2, CO, NO ...) impliquent des métalloporphyrines (molécules formées par la combinaison d'un métal et d'une porphyrine, molécules à structures cycliques impliquées dans le transport de l’oxygène).
Ce transport a lieu par fixation réversible de ces molécules sur le métal du site actif d’une hémoprotéine : l’hème, c'est-à-dire sur la protoporphyrine IX du fer. Cela fait intervenir des processus de ligandation et déligandation complexes résultant d’un équilibre subtil entre l’attraction métal–ligand, et l’attraction et la répulsion du ligand avec l’environnement de l’hème. La contribution relative de ces interactions est mal connue, d’où de nombreuses mesures sur l’énergétique des liaisons métal/ligand. Nombre d’entre elles sont issues de mesures de constantes d’équilibre en phase liquide qui ne permettent pas une comparaison directe avec des calculs théoriques. Cette limitation empêche la compréhension véritable du processus ligandation-déligandation.
Les mesures en phase gazeuse sont très appropriées à l’étude du processus primaire de ligandation/déligandation. Cela a motivé une collaboration, via les appels d’offre nationaux évoqués plus haut, puis largement poursuivie. La dynamique des états excités de ces systèmes a été étudiée sur un modèle : la tétraphényl porphyrine du ruthénium II liée à CO, excitée dans la bande de Soret à 400 nm par un laser femtoseconde. Cela a permis d’explorer la dynamique d’expulsion du ligand, via un état à transfert de charge porphyrine→métal auquel le système accède en 70 fs.
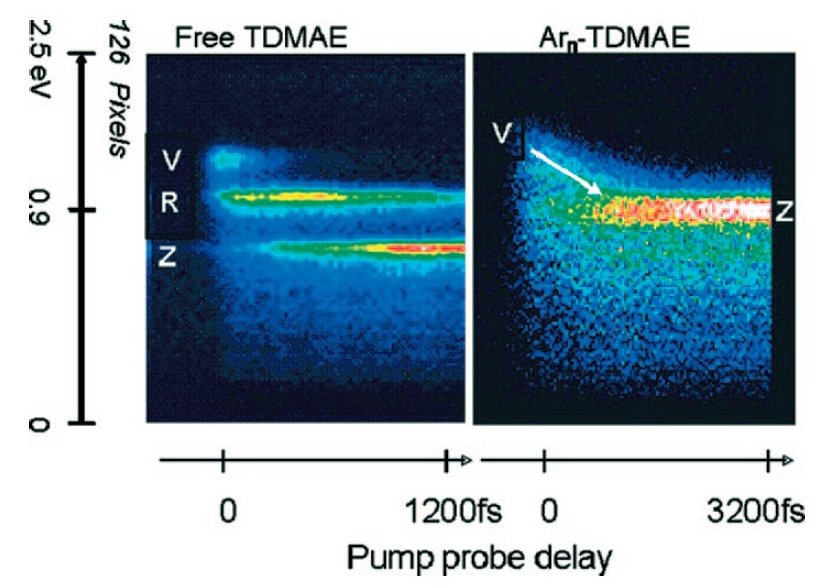
Image synthétique montrant un spectre de photoélectrons en fonction du délai pompe-sonde dans une expérience de femtochimie sur TDMAE libre ou déposé sur agrégat d’argon. L’impulsion pompe excite l’état V, la sonde ionise les états V, R et Z et produit les photoélectrons observés
Une avancée de la femtochimie en phase gazeuse est l’exploration des processus de relaxation dans les états excités des molécules de grande taille. Ceux-ci convertissent l’énergie électronique initiale en vibration intramoléculaire, ce qui assure la stabilité des molécules vis-à-vis de la lumière. Des croisements (coniques) entre surfaces de potentiel, rendent la relaxation rapide et efficace.
Nous avons examiné cette situation sur le tetrakis(dimethyl-amino) éthylène (TDMAE) qui possède une double liaison C=C : l’état excité V(ππ*) centré sur cette liaison se relaxe en moins de 500 fs vers un état excité zwitterionique C+C- (Z). La disparité de forme entre les orbitales V et Z pose le problème de l’origine d’une relaxation aussi rapide. La partie gauche de la figure montre qu’en fait, le transfert V→Z n’est pas direct. Un état de Rydberg (R) sert d’adaptateur entre V et Z. Plus récemment, nous avons examiné cette relaxation dans une phase condensée, simulée par un agrégat d’argon. La partie droite de la figure montre que pendant 1 ps environ, il y a disparition de la structure en trois états V, R, Z et que la structure électronique du système molécule-agrégat évolue continument au cours du temps.
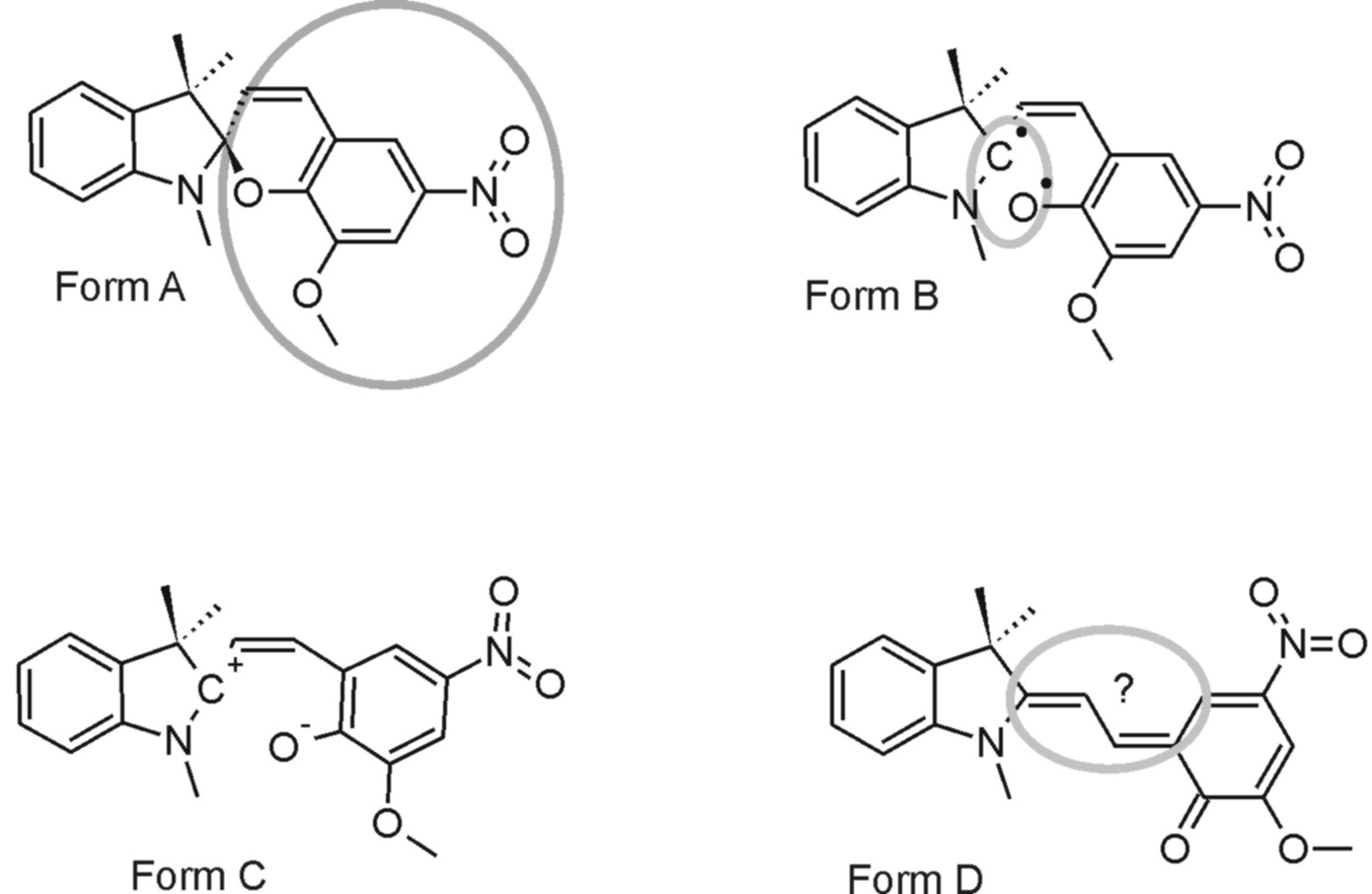
Les spiropyranes et les spirooxazines sont deux classes de molécules photochromes dont les applications commerciales sont considérables. Par exemples, les spirooxazines sont utilisées dans les matériaux actifs à la lumière solaire et, plus encore, comme mémoire et comme interrupteur optique.
Ces molécules possèdent deux isomères dont les propriétés électroniques sont très différentes. Le point clé, responsable de leurs propriétés photochromiques, est la commutation d’un isomère à l’autre par absorption lumineuse. Le mécanisme de cette commutation est encore largement incompris malgré un grand nombre d’études en phase condensée. Il manque une information essentielle : le comportement propre de ces molécules après excitation lumineuse, hors solvatation.
Le groupe de G. Buntinx (CERLA, Villeneuve d’Ascq) est spécialisé dans l’étude de ces molécules en phase condensée. Une collaboration a été entamée avec lui pour étudier la dynamique femtoseconde de telles molécules, isolées en phase gazeuse. Le but est d’en connaitre la dynamique intrinsèque, sans perturbation par un solvant. Une dynamique impliquant plusieurs formes transitoires a été observée (voir figure). On constate des réarrangements structuraux considérables. Une telle complexité, attendue sur le plan théorique, n’avait encore reçu aucune confirmation expérimentale. L’intérêt porté à ce travail nous a permis d’adhérer au GDR International PHENICS (PHoto-switchablE orgaNIC molecular systems & deviceS).
