Shooting the movie of attosecond photoemission
In 1905, Einstein’s theoretical interpretation of the photoelectric effect (the emission of an electron following the absorption of a quantum of light, the photon) revolutionized physics. Because of its extreme speed, this fundamental process was long considered instantaneous. It’s only in the last ten years or so that the development of ultra-short light sources and attosecond metrology (1 as= 10-18 s) has enabled us to gain access to the temporal aspect of this ultra-fast process. Until now, the spatial aspect was not resolved in real time.
An experiment carried out on the ATTOLab platform at CEA Paris-Saclay by a French collaboration comprising researchers from the ATTO group at CEA, CNRS, Université Paris-Saclay, Sorbonne Université and Université Lyon 1 has made it possible for the first time to reconstruct the three-dimensional film of a photoemission process, at the atomic level and on the attosecond scale.
As photoemission is also the basis for some of the finest spectroscopic analysis methods, this work opens the way to a deeper understanding of electronic correlation effects in matter, from atoms and molecules to solids, at work in chemical reactions in particular.
Publication
“Anisotropic dynamics of two-photon ionization: An attosecond movie of photoemission“
A. Autuori, D. Platzer, M. Lejman, G. Gallician, L. Maëder, A. Covolo, L. Bosse, M. Dalui, D. Bresteau, J.-F. Hergott, O. Tcherbakoff, H.J.B. Marroux, V. Loriot, F. Lépine, L. Poisson, R. Taïeb, J. Caillat and P. Salières, Science Advances 8(12) (2022).
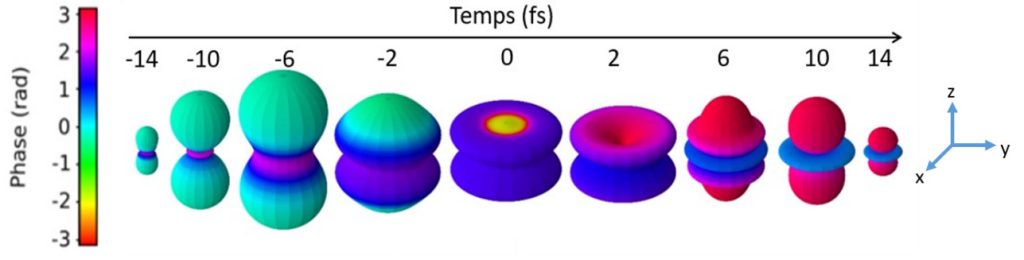
3D movie of the photoemission of a helium atom following the simultaneous absorption of an extreme ultraviolet (XUV) photon and an infrared (IR) photon. The instant t = 0 is placed at the maximum overlap of the XUV IR pulses.
Each instant represents the complex amplitude of the ionization rate
(modulus in polar coordinates, phase given by color code)
How can we see resonances building up in real time?
Spectroscopy has taught us how the very precise measurement of the spectral shape of resonance lines (known as a ‘line profile’) can be used to understand the structure of matter. However, as a time-integrated measurement, spectral lines give only indirect information about the underlying electronic dynamics. The width of the resonance can be related to the time scale of electronic excitation and relaxation, but in the general case this is not sufficient to access the details of the complete dynamics, which must be recovered from advanced modeling.
A typical case is that of self-ionizing resonances, where the system (atom, molecule, nanostructure) can either be ionized directly in the continuum, or be trapped in a highly excited state for a very short time (femtosecond) before reaching the continuum. The interference between the two channels results in an asymmetric line profile, known as the Fano profile, after the Italian theorist Ugo Fano who first modeled this process.
Read more
While the Fano profile has proved extremely effective in analyzing resonance lines measured in a wide variety of systems, the details of how the process unfolds in time have remained elusive, as the ultra-short time scale involved precludes direct time-domain studies.
In collaboration with two theoretical groups (LCPMR-Paris and UAM-Madrid), we have developed a new technique called “Rainbow RABBIT” based on multicolor coherent ionization by a train of attosecond pulses precisely synchronized with an infrared laser pulse. This technique gives direct access to the spectral amplitude and phase of the resonant electron wave packet emitted into the continuum.
A Fourier transform then allows us to resolve the ultrafast time construction of the self-ionizing resonance, revealing the decomposition of the process into two almost consecutive stages governed by quite different time scales (see Figure): during the first 3 femtoseconds, the direct ionization channel dominates; then, the resonant channel begins to contribute as the doubly excited state empties into the continuum, leading to interference between the two channels that eventually shapes the Fano profile.
This technique opens up multiple possibilities for studying highly correlated ultrafast dynamics in a variety of systems, from molecules and nanostructures to surfaces, and for monitoring changes in matter at the most fundamental level.
Publications :
Attosecond dynamics through a Fano resonance: monitoring the birth of a photoelectron
Gruson V., Barreau L., Jiménez-Galan Á., Risoud F., Caillat J., Maquet A., Carré B., Lepetit F., Hergott J.-F., Ruchon T., Argenti L., Taïeb R., Martín F., Salières P.,. Science 354(6313), 734-8 (2016).
Time-frequency representation of autoionization dynamics in helium
Busto D., Barreau L., Isinger M., Turconi M., Alexandridi C., Harth A., Zhong S., Squibb R.J., Kroon D., Plogmaker S., Miranda M., Jimenez-Galan A., Argenti L., Arnold C.L., Feifel R., Martin F., Gisselbrecht M., L’Huillier A., Salières P., J. Phys. B 51, 044002 (2018).
Associated projects: ANR-CIMBAAD coordinated by Pascal Salières, and ITN-MEDEA
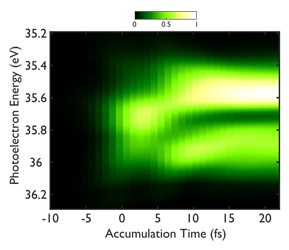
Opposite, the experimental reconstruction of the time construction of the Helium 2s2p self-ionizing resonance in the photoelectron spectrum.
Attosecond photoemission in the inner layer
During the photoemission process, photoelectron scattering on the ionic potential induces tiny ionization delays that provide a very fine probe of the potential, its anisotropy and resonant states.
The recent development of attosecond spectroscopy techniques has made it possible to measure these delays with great precision when electrons are emitted from valence layers, which are relatively easy to ionize.
Read more
Very few measurements have been made in the inner layer, where high photon energies are more difficult to generate. Yet the photoemission of core levels offers a number of advantages.
Firstly, they represent a source of electrons localized to a particular atom in a molecule, unlike valence electrons, which come from delocalized orbitals giving only averaged information on molecular potential.
In addition, inner-layer photoemission is chemically selective, ‘targeting’ the absorption threshold of a specific element, such as ionized iodine in layer 4d.
In this way, it becomes possible to measure the influence on ionization times of different functional groups in an iodine-containing molecule, and thus answer questions such as :
– Does an iodine 4d electron take “longer” to ionize in ICl or CH3I?
– What influence does the grouping (e.g. -Cl or -CH3) have on the path and temporal dynamics of the emitted electron?
Finally, the excited states of matter produced by inner-layer excitation are highly ephemeral, lasting only a few femtoseconds before de-excitation (e.g. Auger), and open up new possibilities in terms of observable phenomena.
Associated projects: PALM MOCAPI, EC-IEF TD-PICO-MF
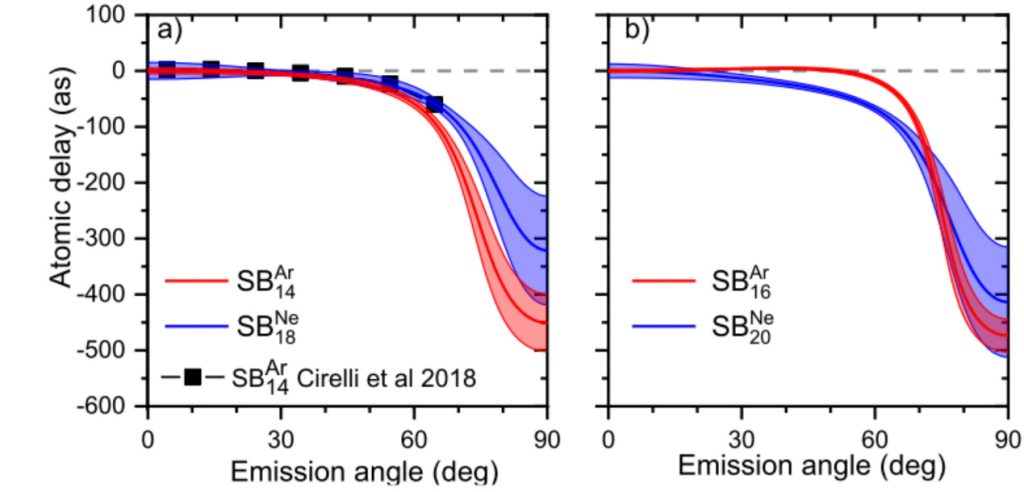
Measuring ionization delays using coincidence spectroscopy
Attosecond photoionization delays are strongly dependent on the potential of the molecule/atom under consideration. An important piece of information is therefore the angular dependence of these delays.
One method of choice for dealing with angularly resolved photoionization is the use of vector correlation spectroscopy in a COLTRIMS-type spectrometer. When molecular ionization is dissociative, this spectroscopy enables the polar emission pattern of electrons in the molecular frame of reference to be recovered to a few approximations.
Read more
The main difficulty associated with the technique is the requirement to be in the “coincidence” regime, i.e. much less than one molecule of interest dissociates per laser shot, enabling a specific electron to be associated with an ionic fragment. This makes for long, delicate experiments.
On the ATTOLab FAB10 line, we were able to implement the RABBIT technique while detecting coincidence photoionization in a COLTRIMS.
This research was coordinated by Danielle Dowek’s group at ISMO (CNRS-Université Paris-Saclay).
This work has extended the theoretical framework of RABBIT to angularly resolved studies, established benchmark data on photoionization times for noble gas atoms, and finally laid the foundations for future comprehensive studies of molecular ionization.
Publication:
First complete polarimetry of high-order harmonics
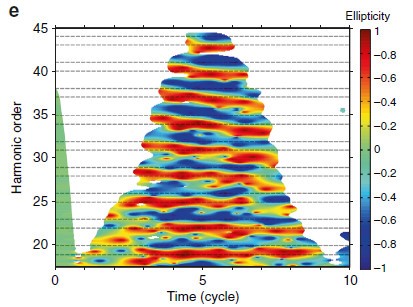
Simulation of HHG induced by ultrashort bichromatic countercircular fields: time-frequency representation of harmonic ellipticity, showing the alternating helicity of 3q 1 and 3q 2 harmonics, but also their temporal inhomogeneity leading to significant depolarization.
Circularly polarized light is a remarkable probe of chirality and magnetization. The advent of ultrashort pulses in the extreme ultraviolet (XUV) and soft X-ray ranges produced by high-order harmonic generation (HHG) has opened up the prospect of studying these phenomena at the natural (sub-) time scales of electronic dynamics and magnetic interactions with atomic selectivity.
Read more
However, two major problems had to be solved:
- i) how to efficiently produce circularly polarized HHG, and
- ii) how to fully characterize its polarization state.
With regard to point ii), complete polarimetry is particularly difficult in the XUV range, where most materials absorb and scatter radiation strongly, and where broadband quarter-wave blades are not available. Research into the polarization properties of harmonic emission began 20 years ago [Weihe et al, PRA 1995], but until now all measurements were incomplete and had to be based on the assumption that XUV light was fully polarized.
The “molecular polarimetry method”, developed by Danielle Dowek’s group (ISMO-Université d’Orsay) on the SOLEIL synchrotron [Veyrinas et al, PRA 2013], is based on the dissociative ionization of NO molecules induced by XUV light. By coincidentally measuring the electron and N ion emitted in a COLTRIMS-type reaction microscope, we determine the angular distributions of photoelectrons in the molecular frame of reference (MFPAD) that simultaneously contain the three Stokes parameters of XUV light.
This enables complete characterization of the polarization state, with direct access to the absolute value and sign of the ellipticity, as well as the degree of polarization of the individual harmonic orders. Applying this technique to the 1 kHz harmonic source at CEA-Saclay was a real experimental challenge, requiring acquisition times of up to 3 hours.
With regard to point i), we investigated two schemes for producing highly elliptical XUV light: HHG in SF6 molecules induced by an elliptically polarized laser [Veyrinas et al, Faraday Discussion 2016], and HHG in argon induced by two circularly polarized counter-rotating laser fields at the fundamental (w) and second harmonic (2w) frequencies of the laser [Barreau et al, Nature Communications 2018]. The first scheme produced high ellipticities, but only for 2 harmonic orders, and with low conversion efficiency.
The real surprise came from the second scheme, which had recently been revived [Fleischer et al., Nature Photon. 2014] due to its potential to efficiently produce circularly polarized XUV emission extending to X-rays [Fan et al., PNAS 2015]. The 3q 1 (3q 2) harmonic orders were assumed to be circularly polarized like the w (2w) field, respectively. Our full characterization revealed unexpected features, such as significant depolarization as well as deviations from circularity.
In particular, it was generally considered that, under “non-pathological” generation conditions, the coherence of the HHG process resulted in fully polarized emission. We have carried out a theoretical study demonstrating that ultrashort generating fields and ultrafast ionization lead to dynamic symmetry breaking, resulting in depolarization and deviations from circularity (see Figure).
The importance of these results is manifold: from a fundamental point of view, they enable a more complete understanding of the HHG process and its polarization properties; from an applied point of view, they pave the way for an optimized and well-characterized source of circularly polarized ultrashort XUV radiation, of interest to a wide community studying ultrafast dichroisms, from solid-state physicists to chemists.
Publications:
“Evidence of depolarization and ellipticity of high harmonics driven by ultrashort bichromatic circularly-polarized fields“
L. Barreau, K. Veyrinas, V. Gruson, S. J. Weber, T. Auguste, J.-F. Hergott, F. Lepetit, B. Carré, J.-C. Houver, D. Dowek, P. Salières, Nature Communications 9, 4727 (2018).
“Molecular frame photoemission by a comb of elliptical high-order harmonics:a sensitive probe of both photodynamics and harmonic complete polarization state.”
K. Veyrinas, V. Gruson, S. J. Weber, L. Barreau, T. Ruchon, J.-F. Hergott, J.-C. Houver, R. R. Lucchese, P. Salières, D. Dowek, Faraday Discussions 194, 161 (2016).