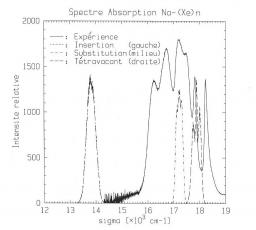
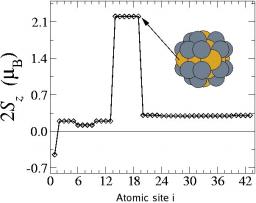
Simulation Monte carlo de l’auto-organisation en étoile de copolymères diblocs en solution
L'ensemble des recherches effectuées à l'IRAMIS s'appuient à la fois sur de l'expérimentation dans le domaine de la physique et/ou de la chimie et sur des développements théoriques importants. Cet ensemble théorique (théorie exacte, approche non linéaire, modélisation, simulation, etc...) est à la base de la compréhension des phénomènes, permet la comparaison avec l'expérience, la validation de modèles et soutient l'ensemble des travaux menés. Les principaux domaines abordés sont les suivants :
- chimie théorique (calculs ab-initio, dynamique moléculaire, effets relativistes dans les éléments f)
- physique de l'irradiation (création de défauts, dépôt d'énergie, …)
- physique des plasmas (haute densité électronique, très haute énergie laser, …)
- physique du solide et physique quantique
- physique statistique (chaos, non linéarité, désordre, …)
- physico-chimie (agrégats, matière molle, solutions, …)
- dynamique réactionnelle.