






2009

« Back to the LENSIS Group page « Back to the Oxides page
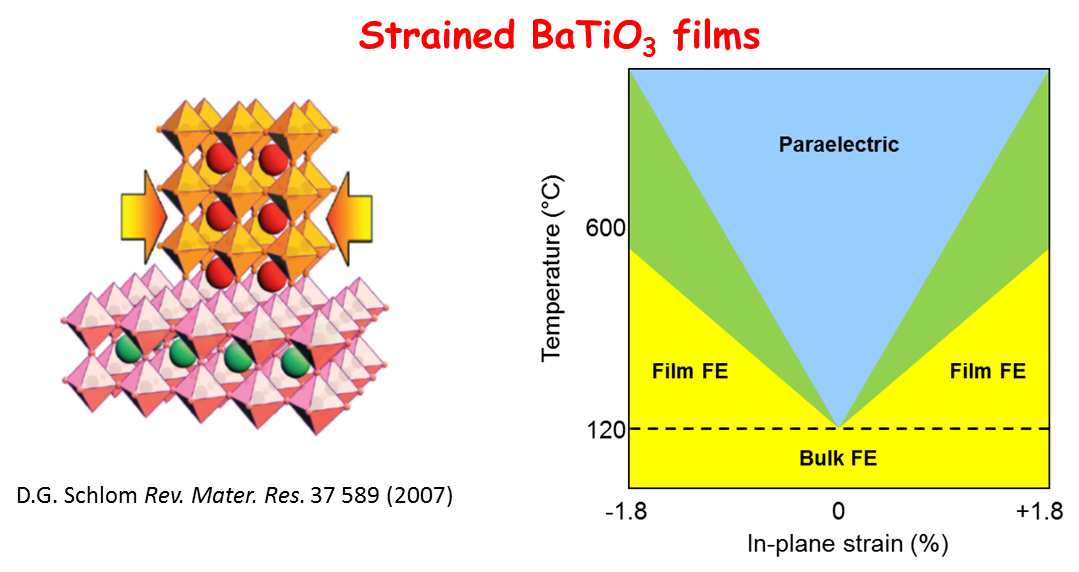
The advent of high quality epitaxial film growth, decisive theoretical advances and experimental tools capable of characterizing in detail ferroelectric materials has led to a resurgence of interest, in particular the perspective of engineering films for ferroelectric-based electronics. Substrate-imposed strain, for example, can increase the Curie temperature, stabilizing ferroelectricity in BaTiO3 up to 600°C.
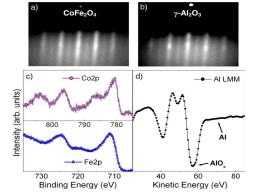
We report for the first time the direct measurement of spin polarization in epitaxial CoFe2O4 tunnel barriers using the Meservey-Tedrow technique. We further analyze the effect of oxidation conditions during film growth on the polarization of the tunneling current (PSF), revealing an important role played by oxygen vacancies in the spin-filter efficiency of this material.
These spin filtering measurements have been performed in collaboration with the team of J. S. Moodera from the Francis Bitter Magnet Lab, MIT.

Head: Cécile Reynaud
This topic is in the field in full rise of nanoscience and nanotechnology, which involves the study of structures or materials which have at least one characteristic dimension below 100 nm and have, therefore, specific properties. We develop our own nano-objects synthesis methods with a bottom-up approach. The aim is the formation of new entities with controlled physical and chemical properties. We study their growth process and their specific properties, particularly those induced by size effects. Taking advantage of the diversity of nanostructured objects that we are able to synthesize (nanoparticles, carbon nanotubes, membranes, electrodes...), we develop original nanomaterials or nanocomposites. Dispersion and suspension of these nano-objects, with or without prior functionalization, are studied in order to enable their convenient and safe manipulation. Many characterization methods are implemented, in particular optical spectroscopy and electron microscopy, often through collaborations. The concerned applications are in the fields of optics, optoelectronics, catalysis, sensors and markers (fluorescent or magnetic), new technologies for energy, new technologies for health, next generation nuclear materials, etc. This activity is in close relation with the national and international network of basic and applied research through many national and international contracts.
Introduction
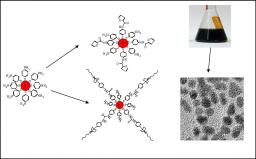
This research deals with the elaboration and study of solid state nanocomposite structures built using pre-synthesized objects handled in liquids (bottom-up approach). This approach affords a way to study the impact of nanoscale modifications on the macroscopic properties of nanostructures which can be therefore optimized. It is developed on model systems based on platinum nanoparticles chemically functionalized with 4-mercaptoaniline (mother particles) from which chemical modification performed on this organic crown affords new nano-objets with controlled features and composition. Thanks to the organic crown grafted on the platinum core (2nm diameter), the different nanoparticles can be handled in solution like molecules.
The room-temperature fluorescence properties of DNA nucleoside and nucleotide aqueous solutions are studied by steady-state and time-resolved spectroscopy. The steady-state fluorescence spectra, although peaking in the near-UV region, are very broad, extending over the whole visible domain. Quantum yields are found to be mostly higher and the fluorescence decays faster than those reported in the literature. The fluorescence spectra of the 2‘-deoxynucleosides are identical to those of the 2‘-deoxynucleotides, with the exception of 2‘-deoxyadenosine, for which a difference in the spectral width is observed. The steady-state absorption and fluorescence spectra do not show any concentration dependence in the range 5 × 10-6 to 2 × 10-3 M. All fluorescence decays are complex and cannot be described by monoexponential functions. From the zero-time fluorescence anisotropies recorded at 330 nm, it is deduced that after excitation at 267 nm the largest modification in the electronic structure is exhibited by 2‘-deoxyguanosine. In the case of purines, the fluorescence decays and quantum yields are the same for 2‘-deoxynucleosides and 2‘-deoxynucleotides. In contrast, for pyrimidines, the fluorescence quantum yields of nucleotides are higher and the fluorescence decays slower as compared to those of the corresponding nucleosides showing that the phosphate moiety affects the excited-state relaxation.
The first comprehensive quantum mechanical study of solvent effects on the behavior of the two lowest energy excited states of uracil derivatives is presented. The absorption and emission spectra of uracil and 5-fluorouracil in acetonitrile and aqueous solution have been computed at the time-dependent density-functional theory level, using the polarizable continuum model (PCM) to take into account bulk solvent effects. The computed spectra and the solvent shifts provided by our method are close to their experimental counterpart. The S0/S1 conical intersection, located in the presence of hydrogen-bonded solvent molecules by CASSCF (8/8) calculations, indicates that the mechanism of ground-state recovery, involving out-of-plane motion of the 5 substituent, does not depend on the nature of the solvent. Extensive explorations of the excited-state surfaces in the Franck−Condon (FC) region show that solvent can modulate the accessibility of an additional decay channel, involving a dark n/π* excited state. This finding provides the first unifying explanation for the experimental trend of 5-fluorouracil excited-state lifetime in different solvents. The microscopic mechanisms underlying solvent effects on the excited-state behavior of nucleobases are discussed.
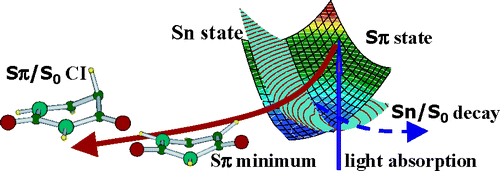
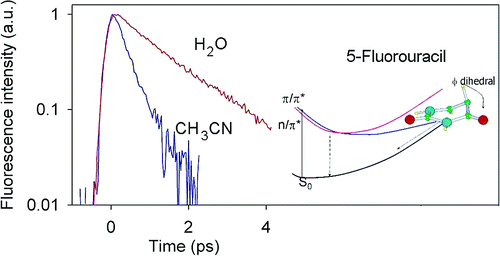
The excited-state dynamics of 5-fluorouracil in acetonitrile has been investigated by femtosecond fluorescence upconversion spectroscopy in combination with quantum chemistry TD-DFT calculations ((PCM/TD-PBE0). Experimentally, it was found that when going from water to acetonitrile solution the fluorescence decay of 5FU becomes much faster. The calculations show that this is related to the opening of an additional decay channel in acetonitrile solution since the dark n/π* excited state becomes near degenerate with the bright π/π* state, forming a conical intersection close to the Franck−Condon region. In both solvents, a S1−S0 conical intersection, governed by the out-of-plane motion of the fluorine atom, is active, allowing an ultrafast internal conversion to the ground state.
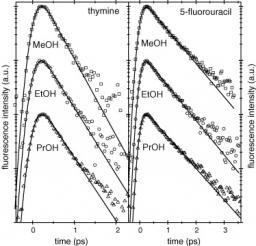
The excited state properties of uracil, thymine and four analogous uracil compounds have been studied in acetonitrile by steady-state and time-resolved spectroscopy. The excited state lifetimes were measured using femtosecond UV fluorescence upconversion. The excited state lifetimes of uracil and its 1- and 3-methyl substituted derivatives are well described by one ultrafast (100 fs) component. Five substituted compounds show a more complex behavior, exhibiting longer excited state lifetimes and bi-exponential fluorescence decays. These longer decays are substantially faster in acetonitrile than in aqueous solution showing that the excited state deactivation mechanism is in part governed by the solvent.
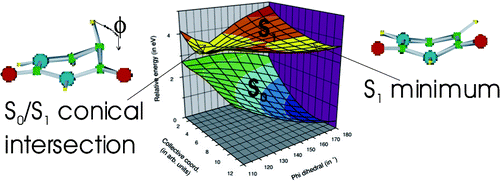
The excited-state properties of uracil, thymine, and nine other derivatives of uracil have been studied by steady-state and time-resolved spectroscopy. The excited-state lifetimes were measured using femtosecond fluorescence upconversion in the UV. The absorption and emission spectra of five representative compounds have been computed at the TD−DFT level, using the PBE0 exchange-correlation functional for ground- and excited-state geometry optimization and the Polarizable Continuum Model (PCM) to simulate the aqueous solution. The calculated spectra are in good agreement with the experimental ones. Experiments show that the excited-state lifetimes of all the compounds examined are dominated by an ultrafast (<100 fs) component. Only 5-substituted compounds show more complex behavior than uracil, exhibiting longer excited-state lifetimes and biexponential fluorescence decays. The S0/S1 conical intersection, located at CASSCF (8/8) level, is indeed characterized by pyramidalization and out of plane motion of the substituents on the C5 atom. A thorough analysis of the excited-state Potential Energy Surfaces, performed at the PCM/TD−DFT(PBE0) level in aqueous solution, shows that the energy barrier separating the local S1 minimum from the conical intersection increases going from uracil through thymine to 5-fluorouracil, in agreement with the ordering of the experimental excited-state lifetime.
Cytosine methylation, which determines the hot spots for DNA photo-damage, is shown to induce a red-shift of the nucleoside absorption spectrum, making the chromophore more vulnerable to solar radiation, and a tenfold increase of the fluorescence lifetime, making excited state reactions more probable. A femtosecond investigation of the excited state deactivation reveals a quite complex mechanism.
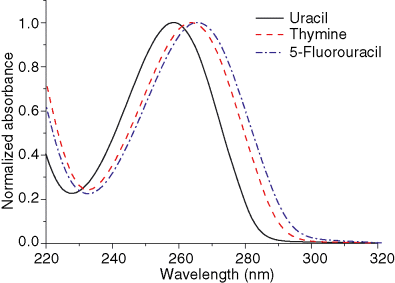
We report a comparison of the steady-state absorption and fluorescence spectra of three representative uracil derivatives (uracil, thymine and 5-fluorouracil) in alcoholic solutions. The present results are compared with those from our previous experimental and computational studies of the same compounds in water and acetonitrile. The effects of solvent polarity and hydrogen bonding on the spectra are discussed in the light of theoretical predictions. This comparative analysis provides a more complete picture of the solvent effects on the absorption and fluorescence properties of pyrimidine nucleobases, with special emphasis on the mechanism of the excited state deactivation.
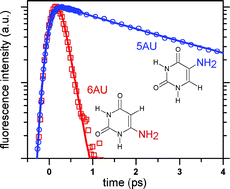
The excited state deactivation of two amino-substituted uracils, 5-aminouracil (5AU) and 6-aminouracil (6AU) in aqueous solution was studied by femtosecond fluorescence upconversion. The fluorescence of 6AU decays as fast as that of uracil with a unique time constant of about 100 femtoseconds. The fluorescence of 5AU exhibits a more complex behavior, fundamentally different from what we found in any other uracils: the decays are globally slower (up to several picoseconds) and depend strongly on the wavelength. This difference is attributed to the particular character of the amino group, affecting the out-of-plane motion of the 5-substituent which has been shown to be crucial for the ultrafast internal conversion occurring in uracils. Our observations indicate instead the formation of a transient fluorescent state which in turn is deactivated by a different relaxation process specific to the amino group
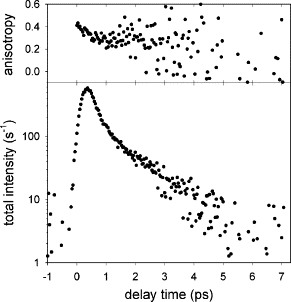
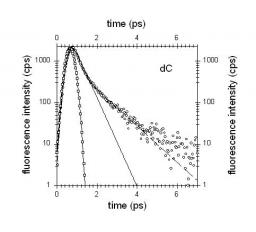
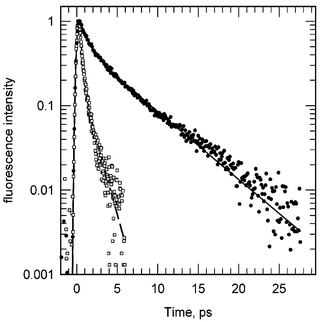
We report a femtosecond spectroscopic study of the DNA base cytosine in aqueous solution at room temperature. Two different experimental techniques were used, fluorescence upconversion and transient absorption, providing complementary information on the excited state relaxation. While the fluorescence decay is clearly bi-exponential, with an ultrafast (0.2 ps) and a slower (1.3 ps) component, the decay of the transient absorption signal is mono-exponential with a 1.1 ps characteristic time. In addition, the fluorescence anisotropy is also found to decay in a bi-exponential manner. The results are discussed in terms of possible non-radiative relaxation processes that may intervene in the deactivation of the excited state.
The excited state lifetimes of uracil, thymine and 5-fluorouracil have been measured using femtosecond UV fluorescence upconversion in various protic and aprotic polar solvents. The fastest decays are observed in acetonitrile and the slowest in aqueous solution while those observed in alcohols are intermediate. No direct correlation with macroscopic solvent parameters such as polarity or viscosity is found, but hydrogen bonding is one key factor affecting the fluorescence decay. It is proposed that the solvent modulates the relative energy of two close-lying electronically excited states, the bright ππ* and the dark nπ* states. This relative energy gap controls the non-radiative relaxation of the ππ* state through a conical intersection close to the Franck–Condon region competing with the ultrafast internal conversion to the ground state. In addition, an inverse isotope effect is observed in D2O where the decays are faster than in H2O.
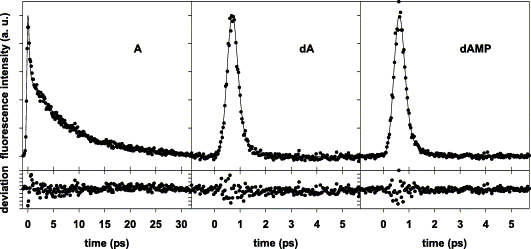
Aqueous solutions of adenine (A), deoxyadenosine (dA) and deoxyadenosine 5′-monophosphate (dAMP) were studied in room temperature by femtosecond fluorescence upconversion. The fluorescence decays cannot be described by single exponentials. They consist of an ultrafast component (230 fs for A, <100 fs for dA and dAMP) and a slower one (8 ps for A, 0.5 ps for dA and dAMP). The slow component constitutes 95% of the total fluorescence (time-integrated) for the base while only 24% for the nucleoside or the nucleotide. The initial fluorescence anisotropy is 0.30±0.03 for A, 0.25±0.05 for dA and dAMP. The anisotropy of the A fluorescence partially decays during its lifetime due to rotational diffusion.
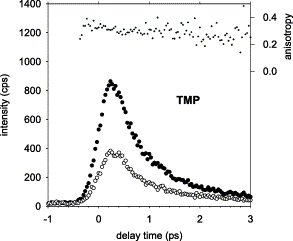
We report fluorescence measurements of DNA components performed on the femtosecond time-scale using the fluorescence upconversion technique. Aqueous solutions of thymine (T), thymidine (dT) and thymidine 5′-monophosphate (TMP) were studied in room temperature by excitation at 267 nm and detection at wavelengths between 310 and 380 nm. The fluorescence decays are complex and cannot be described by single exponentials. About 25% of the fluorescence disappears within 150 fs and the remaining part decays more slowly when going from the base through the nucleoside to the nucleotide. The initial fluorescence anisotropy was found to be 0.35±0.03 and did not show any drastic change on the examined time interval.
Photoinduced biological processes are complex and can often be reduced to a series of sequential events after the absorption of the photon. Each of these steps, if separable, can be compared with the evolution of a much simpler system, mimicking its essential characteristics, a biomimetic system. This stems from the very local properties of the initial event where a small reaction centre has been excited, spatially limited within the biomolecule. Gas phase conditions provide the unique way to study these model systems.
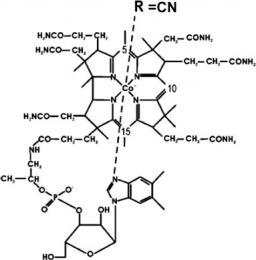
The time evolution of electronically excited vitamin B12 (cyanocobalamin) has been observed for the first time in the gas phase. It reveals an ultrafast decay to a state corresponding to metal excitation. This decay is interpreted as resulting from a ring to metal electron transfer. This opens the observation of the excited state of other complex biomimetic systems in the gas phase, the key to the characterisation of their complex evolution through excited electronic states.

Bertrand Carré
Head of Attophysic group
CEA- Saclay
DSM/IRAMIS
Service of Photons, Atoms and Molécules
Bât. 522 p. 113
91191 Gif sur Yvette Cedex
France
Tél : +33 1 69 08 58 40
Fax : +33 1 69 08 12 13
e-mail : bertrand.carre@cea.fr
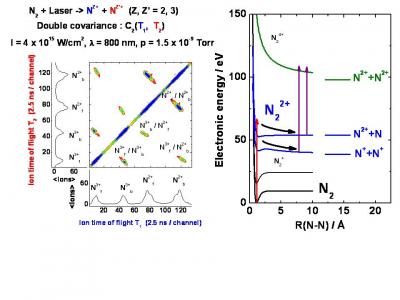
Whereas tunnel single ionization is an attosecond process taking place within a fraction of an optical cycle, multiple ionization occurs within at least one half-cycle in the case of non sequential multiple ionization or within several cycles. In molecules, multiple ionization leads to the fragmentation into multicharged atomic fragments and is usually labelled Coulomb explosion. In principle Coulomb explosion allows one to image the positions of the atoms within the molecule provided that multiple ionization times remain small in comparison with the timescales of nuclear motions. In these experiments, the time resolution is a major issue in order to compete with accelerator-based techniques such as foil-induced and ion-induced multiple ionizations which occur during a few tens of attoseconds. The advantage of the laser excitation lies in the compactness of the experimental set-ups and more importantly in the possibility to perform pump-probe excitation schemes for imaging excited molecules. Using few-cycle laser pulses to multiply ionize molecules, we study the simultaneous electronic and nuclear relaxations as a powerful probe of the ultrafast ionization sequence at the femtosecond time scale. The molecular systems range from the hydrogen molecule to simple polyatomic molecules. The molecular response is analysed using the multicharged atomic fragments time-of-flight detection, ions correlations, energy analysis, and fluorescence detection.
Thomas-Fermi theory of multiple ionization
Main investigator: M. Brewczyk (University of Bialystok, Poland)
Molecular multiple ionization is studied using a hydrodynamic model which allows one to deal with many-electron systems in intense laser fields. The predicted kinetic energy releases of the fragmentation channels are in good agreement with the measured energies in CO2 and N2O.
Ph. Hering, Thèse de doctorat, Paris (1999)
Ph. Hering, M. Brewczyk, and C. Cornaggia, Phys. Rev. Lett. 85, 2288 (2000)
The project proposes to develop a new gas-phase electron diffraction technique using state-of-the-art femtosecond laser technology and the related attosecond physics. In principle, the method allows to analyze structures of transient molecular species with a resolution approaching a few femtoseconds. Nowadays the spectacular progress in laser physics has opened the way to a full control of optical tunnel ionization of atoms and molecules in the attosecond time domain using carrier-envelope-phase-locked few-cycle pulses. Since its proposal in the 1990s by Kuchiev, Schafer and Kulander, and Corkum, the so-called rescattering model has been successful in explaining many new highly non-linear effects such as non sequential double ionization and XUV attosecond pulse generation in terms of quasi-classical orbits of the ionized electron which is driven back onto the ion core by the laser electric field. Following these advances, we propose to control the attosecond dynamics of the first ionization step and the electronic rescattering paths in aligned molecules using carrier-envelope-phase-locked few-cycle laser pulses. The main objective will be to process time-resolved electronic diffraction patterns of molecules using well-established unimolecular experimental diagnostics such as ion and electron spectroscopy. In this scheme, the linearly-polarized laser field drives the corresponding electron flux back onto the ion core. The associated De Broglie wavelength is of the order of internuclear distances thus opening the way to interference effects and structural analysis. Rescattering following tunnel ionization may be elastic or inelastic depending on the energy exchange with the remaining ion core. Our main interest will be focused on elastic rescattering which is the operating physical effect for photoelectron diffraction.
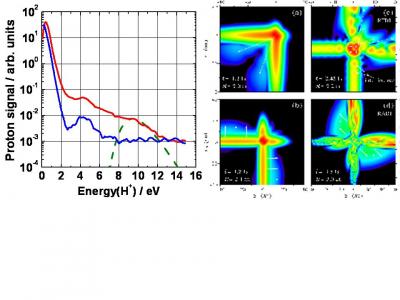
At intensities below 1014 Wcm-2, double ionization of H2 is dominated by recollision excitation of H2+ followed by field ionization of transient excited states. A full quantum non-Born-Oppenheimer model shows that rescattering produces a coherent superposition of excited states which present a pronounced transient H+H- character. This excitation is followed by either field-induced double ionization or the formation of short-lived attosecond autoionizing states. In the first case, electrons are ejected in the same direction. In the second case, electrons are ejected in opposite directions. See the butterfly pattern in the following figure.
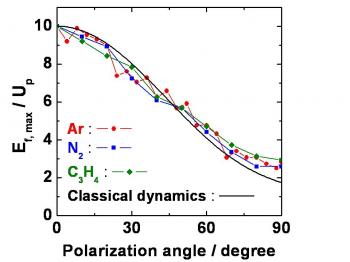
We have implemented pulse compression on the Sofockle laser (800 nm, 600 µJ, 40 fs) after testing different schemes to produce a spatially-homogeneous spectral broadening through self-phase modulation in argon. We obtained pulse durations between 8 and 10 fs, energies up to 200 µJ with intensities up to 1016 Wcm-2.
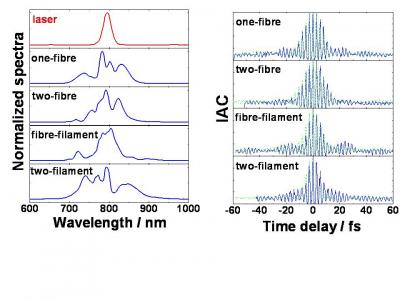
In the 1990s, Coulomb explosion of small molecules has been extensively studied using pulses durations of a few tens of femtoseconds. The stretching of the internuclear separation R was observed during multiple ionization and lead to the important concept of Charge Resonance Enhanced Ionization discovered by A. Bandrauk et al. We have shown that the excitation of N2 or CO2 with 10-fs pulses inhibits any significant stretching. In addition, the fragmentation yields are lower in few-cycle pulses because of the 1/R scaling of the molecular multi-ionization thresholds. These results open the way to femtosecond time-resolved Coulomb explosion imaging of molecules using few-cycle laser pulses.
Time-resolved photoion and photoelectron velocity mapped images from NO2 excited close to its first dissociation limit [to NO(X2P) + O(3P2)] have been recorded in a two colour pump–probe experiment, using the frequency-doubled and frequency-tripled output of a regeneratively amplified titanium–sapphire laser. At least three processes are responsible for the observed transient signals; a negative pump–probe signal (corresponding to a 266 nm pump), a very shortlived transient close to the cross-correlation of the pump and probe pulses but on the 400 nm pump side, and a longer-lived positive pump–probe signal that exhibits a signature of wavepacket motion (oscillations). These transients have two main origins; multiphoton excitation of the Rydberg states of NO2 by both 266 and 400 nm light, and electronic relaxation in the 12B2 state of NO2, which leads to a quasi-dissociated NO2 high in the 12A1 electronic ground state and just below the dissociation threshold.

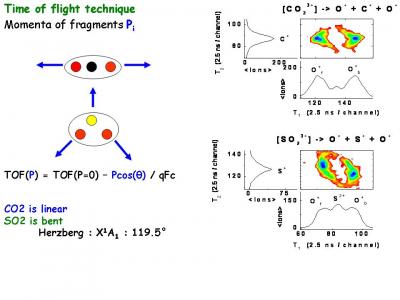
Collaborations : A. Huetz et al., Laboratoire d’Interaction des X Avec la Matière, CNRS Université Paris-Sud (Orsay, France)
Two-color XUV/IR ionization of rare gases has been investigated in so-called “complete experiments”, where all the momenta of electrons and ions from the same ionization event are simultaneously measured (vectorial correlations), using a momentum imaging spectrometer in the coincidence mode (developed by the LIXAM). In single photoionization (PI) of He, angular distribution evidences interferences between open channels [O. Guyétand et al., J. Phys. B 41, 051002 (2008)]. Interestingly, the interference depends on the attosecond delay between harmonics and laser pulses, as illustrated in the simulations in the figure, in good agreement with the delay-averaged experimental points. Conversely, angular distributions in PI can be used to characterize the XUV spectral phase, besides other methods such as RABBITT (see High Harmonic Generation and Attosecond physics).
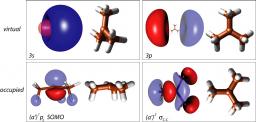
The excited state dynamics of the tert-butyl radical, t-C4H9, was investigated by femtosecond time-resolved photoionization and photoelectron spectroscopy. The experiments were supported by ab initio calculations. Tert-butyl radicals, generated by flash pyrolysis of azotert-butane, were excited into the A 2A1 (3s) state between 347 and 307 nm and the 3p band at 274 and 268 nm and ionized by 810 nm radiation, in a [1+2’] or [1+3’] process. Electronic structure calculations confirm that the two states are of s- and p-Rydberg character, respectively. The carbon framework becomes planar and thus ion-like in both states.
The time evolution of electronically excited vitamin B12 (cyanocobalamin) has been observed for the first time in the gas phase. It reveals an ultrafast decay to a state corresponding to metal excitation. This decay is interpreted as resulting from a ring to metal electron transfer. This opens the observation of the excited state of other complex biomimetic systems in the gas phase, the key to the characterisation of their complex evolution through excited electronic states.
Collaborations : S. Guizard et al., CEA-IRAMIS-Laboratoire des Solides Irradiés (Palaiseau, France)
A. Belski, Laboratoire de Physico-Chimie des Matériaux Luminescents, Université Lyon 1, (Villeurbanne, France)
P. Martin et al., Centre d’Etudes des Lasers Intenses et Applications (Bordeaux, France)
M. Kirm et al., Institute of Physics, University of Tartu (Tartu, Estonia)
A. Vasil’ev, Department of Optics and Spectroscopy, Moscow Lomonosov University (Moscow, Russia)
L. Juha et al., Institute of Physics, Academy of Sciences of the Czek Republic (Prague, Czek Republic)
As compared with IR-visible laser excitation, intense XUV pulses induce specific excitation in solids. The main feature is that single-photon excitation is localized at the surface (absorption length ~ nm), where high density of electronic excitation can be obtained (~1020 /cm3). No further multi-photon heating of the electrons takes place so that the energy is deposited in a controlled manner, in space, time and amount. This type of excitation is particularly relevant for studying relaxation dynamics at femtosecond scale.
As an example, we have investigated the electronic relaxation after XUV excitation in dielectric tungstate crystals, e.g., CdWO4 and CaWO4, which are used as fast scintillators. Long term relaxation – at ns to µs scale – is monitored through the time-resolved luminescence of self-trapped excitons (STE). We evidence that non radiative decay channels can efficiently quench the excitons at short time (< ns) and high excitation density; they involve STE-STE (dipole-dipole) interaction [M. Kirm et al. Physica Status Solidi (c) 4, 870 (2007) ; M. De Grazia et al., Proc. SPIE Vol. 6586, 65860I (2007)].
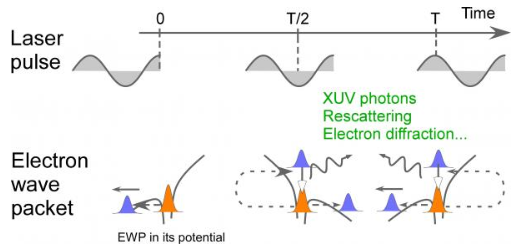
Atoms in a strong laser field: an electron wave packet is launched and driven by the field over one optical cycle. The EWP can return to its parent ion and be scattered as an outgoing electron wave or an attosecond burst of XUV light. The EWP recollision has therefore a double interest: it can be exploited either as a probe of the system with an extreme resolution, or as an ultra-short source of XUV light.
The dynamics of atomic and molecular electrons in a strong laser field is particularly rich and has been a central research topic at LIDYL for more than thirty years. This program is experimentally and theoretically continued in the Attophysics group.
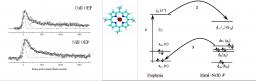
Basically, the ultra-fast electron dynamics in a strong laser field can be described from both quantum and semi-classical concepts, as for instance electron wave packets and electron trajectories, respectively. The semi-classical picture has popularized the elementary dynamical process under the so-called “three-step” model [P. Corkum, Phys. Rev. Lett. 71, 1994 (1993)].
Its profound physical content is illustrated in the figure. When the atom (molecule) is submitted to a laser field – in the intensity range 1013-1016 W/cm2, that is strong enough to distort significantly the core potential -, an electron initially in a valence orbital can escape the core (step 1). This electron subsequently "rides" the laser field and may return back to its parent core within one optical cycle (of duration 2.7 fs = 2.7 10-15 s with the infra-red lasers we use), after it has gained kinetic energy in the field up to a few tens or even hundreds of electronvolts (step 2). In the recollision with the core, the electron can be quasi-elastically scattered (electron diffraction) or inelastically but coherently scattered (step 3). In the latter inelastic recollision, the electron can either further ionize the core or recombine radiatively with it, releasing its energy as an attosecond burst of extreme–UV light. The above three steps including the attosecond emission constitute the elementary sequence in High Harmonic Generation or HHG, first observed in 1987 simultaneously in Chicago and Saclay [A. Mc Pherson et al., J. Opt. Soc. Am. B 4, 595 (1987), M. Ferray et al., J. Phys. B 21, L31 (1988)]. Each optical cycle drives two recollisions so that a train of attosecond pulses in produced in HHG; their temporal characterisation was first achieved in Saclay in 2001 [P.-M. Paul et al., Science 292, 1689 (2001)].
The atomic/molecular electron dynamics in the strong field encompass basic processes, such as ionization and EWP scattering in the different channels, which are studied for themselves along by two research lines. They are detailed in the Multiple ionization & Molecular Imaging and High Harmonic Generation and Attosecond physics pages.
Now, speaking quantum mechanics, the electron is better described as an electron wave packet (EWP) that dynamically splits into two parts, respectively bound and quasi-free, in the laser field, where the quasi-free component undergoes the recollision and scattering onto the core. The free EWP has a de Broglie wavelength in the Angstrom range, which makes it a very appropriate local probe of the system which extends over a comparable scale. Since the EWP probe has attosecond temporal resolution, it can in principle image ultra-fast motion of electrons and nuclei in molecules. Two research lines, described in the Ultra-fast imaging of molecules from electron diffraction and High Harmonic Generation and Attosecond physics pages, build on this "self-probing" paradigm.

Recolliding EWP in a strong laser field. The two coherent scattering channels, EWP diffraction and EWP radiative recombination keep an imprint of the nuclear structure and the electronic orbital in the molecule.
Besides the fundamental studies of the electron dynamics in strong field, and its use as a probe of transient systems, HHG provides with a source of ultra-short coherent pulses in the XUV (from 100 nm down to a few nm). The source's brightness, which reflects the high instantaneous flux and coherence in both the "narrowband" femtosecond and "broadband" attosecond ranges and its natural synchronization with a driving laser, make it very attractive for a number of applications. Among the Examples of applications we have performed multi-color Photoionization in the gas phase, and studies of XUV/solid interaction in the solid state. The coherence properties and partial tunability of the HHG source make it attractive for Seeding a Free Electron Laser, which constitutes another research line. A promising new application concerns the Coherent diffraction imaging of nanometric objects. Most of the applications are developed in collaboration with expert groups, either in France or in Europe, USA, Canada, Japan,…
Eventually, we pursue a theoretical activity to support the several experimental programs. It focuses on microscopic aspects of the gas phase-strong field interaction, i.e., the electron dynamics in atoms and molecules, including Strong Field Approximation (SFA) models in HHG. It also deals with the macroscopic aspects of the interaction, with the development of 3D propagation codes for the laser and XUV fields.
PERPETUALLY UNDER CONSTRUCTION
Interacting magnetic (single-domain) nanoparticles
Single domained ferro- or ferri-magnetic nanoparticles with unique anistropy axis (easy-magnetization axis) are superparamagnetic (SPM) in the absence of inter-particle interactions. That is, the magnetic moment of a particle can fluctuate randomly by thermal fluctuations at high enough temperatures, just as an atomic spin in a paramagnetic material. At low temperatures, the thermal energy becomes smaller than the anisotropy barrier energy inducing particles' magnetic moments to be blocked in the direction of the easy-magnetization axis. This blocking of magnetic moments occur at the temperature TB determined by the particle's size and its composition.
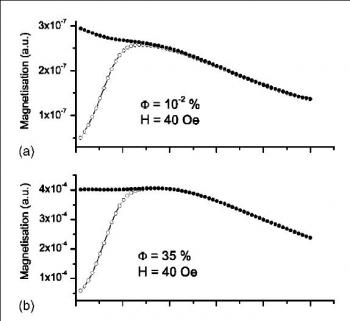
When nanoparticles are sufficiently close to one another, the random, long-range dipole-dipole interactions create a collective phase at low temperatures. Such concentrated nanoparticle assemblies can be made into regular crystal lattice or in a completely random configuration when dispersed in fluid media such as water, oil, or glycerol. In these systems, the dipole-dipole interaction energy added to the individual partciles' anisotropy energy pushes the 'blocking' temperature higher. In some cases of concentrated, monodisperse nanoparticles in frozen media (called ferrofluids) a magnetic state of Superspin Glass has been witnessed. This state is analogous to atomic spin glass states in which the randomness of spin interactions create frustration among them such that a true ground state can never be reached. The name 'superspin' has its origin in the individual nanoparticle's large magnetic moment (e.g. 104 μB per particle of γ-Fe2O3 with 8.6nm diameter). Emblematic sign of spin-glass behavior such as the critical slow down near transition temperature as well as the aging and memory effects (although small) have been observed in frozen ferrofluids.
In our group, we have focused our research effort on the Out-of-Equilibrium dynamics in the superspin glass state of concentrated maghemite ferrofluids. More specifically, we have examined the aging behavior through the thermoremanant magnetization, the AC susceptibility relaxation and zero-field cooled magnetization measurements through which the growing number of correlated superspins has been extracted. Recently, we have investigated the effect of textruization (the anisotropy axis alignment) on the aging dynamics of ferrofluid superspin glass. These experimental studies were conducted using a bulk SQUID magnetometer (CRYOGENIC(TM) S600).
Electronic and chemical structure of functional oxides
We study oxide band structure, surface and interface chemistry, ferroelectric and multiferroic films.
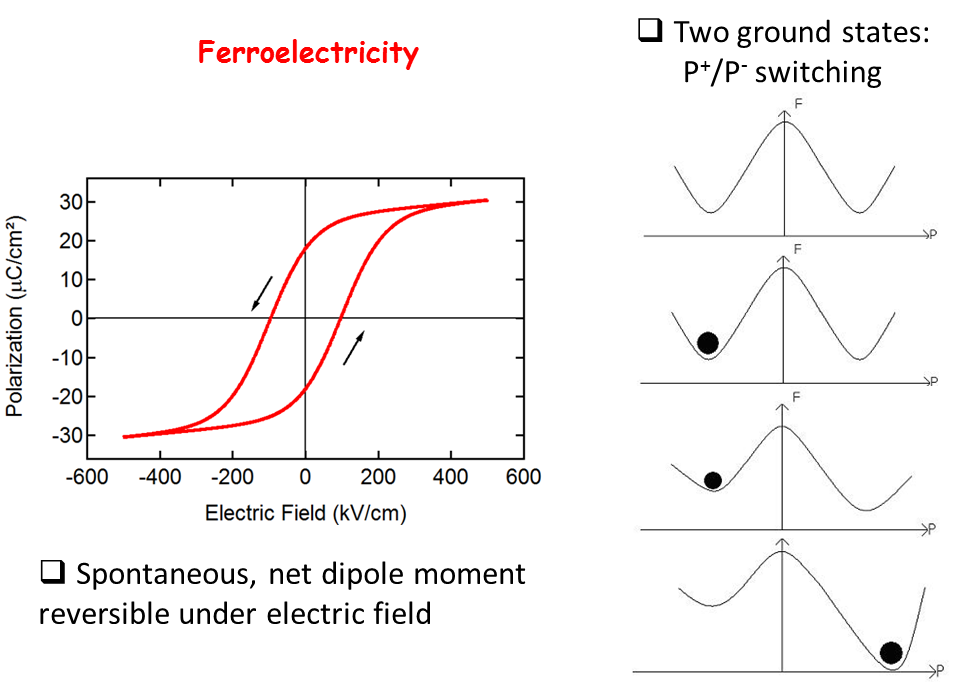
The defining property of a Ferroelectric (FE) material is a spontaneous macroscopic polarization which can be reversed under an applied electric field. The polarization as a function of applied electric field exhibits a hysteresis loop, analogous to ferromagnetic materials, hence the name ferroelectricity.
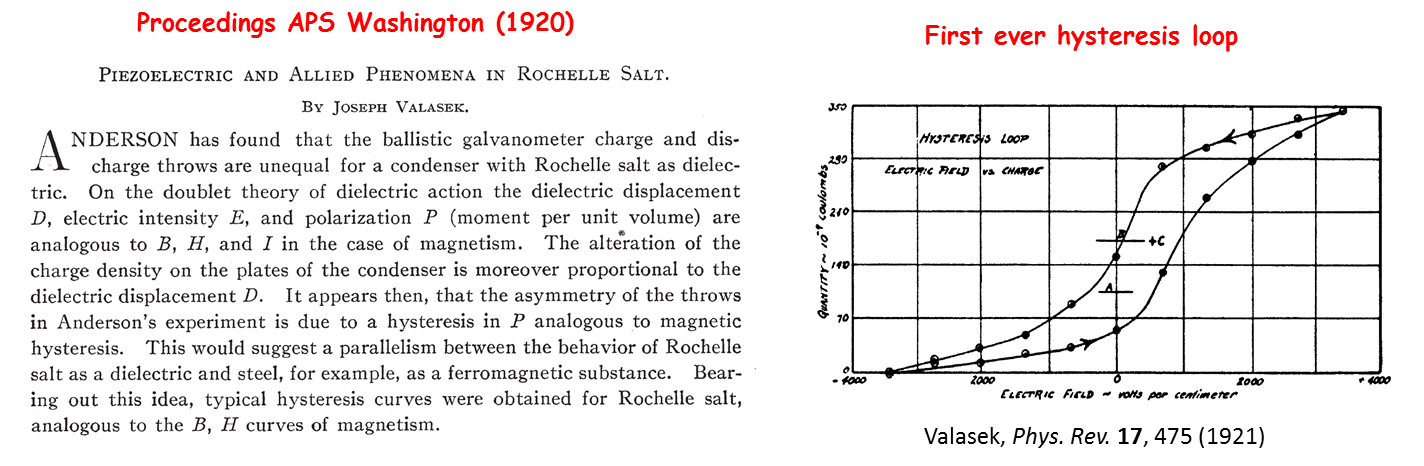
It was discovered by Valasek from the University of Minnesota who presented his results at the Washington meeting of the American Physical Society in April 1920.
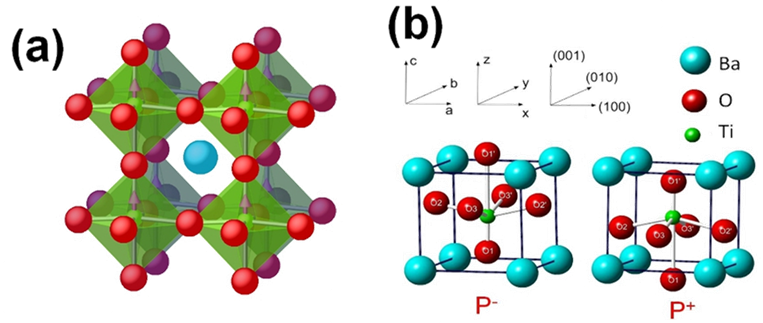
Perovskite oxides, of general formula ABO3 with a pseudocubic structure, where A and B are two different cations, furnish many interesting ferroelectrics. The B-type cation is octahedrally coordinated with oxygen. In the example shown, BaTiO3, it is the relative symmetry breaking displacement of the Ti atoms with respect to the O atoms which is responsible for the spontaneous polarization. BaTiO3 has three ferroelectric phases: tetragonal, orthorhombic and rhombohedral.
« Back to the Group page « Back to the Oxides page
The question of the interface is a key issue in multi-ferroïc heterostructures. Hybridization between filled d orbitals responsible for magnetization and empty d orbitals in the ferroelectric oxide may be one path to such coupling. Several coupling mechanisms have been identified and these can be quite complex. For example, the charge ordering of a magnetic layer can be modulated by the polarization state of an adjacent ferroelectric.
