2006
The study of chemical properties of molecular complexes in solution or at interfaces cannot ignore the quantum nature of electrons. Ab initio molecular dynamics (e.g., of the Car-Parrinello type) must then supersedes classical simulations. Examples of our contributions in this field are the development of pseudopotentials associated with a plane waves basis set and the calculation of free-energy profiles.
Molecular dynamics fails for long time phenomena. At the opposite, pure statistical methods (transition state or RRKM theories) get rid of this problem by phase space volume estimate, but assuming the questionable hypothesis of a fast intra dynamics. We have implemented a mixed statistical dynamical treatment which includes intra dynamics by means of kinetic equations for the states populations. This formalism can be applied either to chemical reactions or to physical chemical processes, such as evaporation or diffusion by jumps.

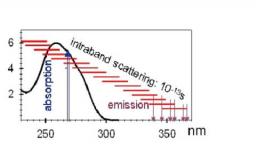
The trapping of molecules in a rare gas matrix is simulated using an original method based on molecular dynamics calculations. Absorption and excited spectra are also simulated.
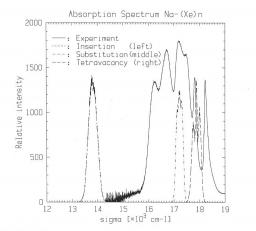
Isolation of atomic sodium in solid host xenon matrices: In collaboration with C. Crépin (LPPM, Orsay), J. G. McCaffrey and M. Ryan (NUI Maynooth, Ireland) a combined theoretical and experimental study is undertaken to characterise the isolation of atomic sodium in solid host xenon matrices. By molecular dynamics (MD) calculations we simulate both the deposition of sodium atom in the xenon matrix and the absorption and emission spectra. Results of deposition show that the sodium atom prefers to occupy a substitution site where only one atom of xenon was removed. Comparison between spectra obtained for different potentials shows the high sensitivity of the method to weak energy variations. MD calculations on the excited state surface allow to obtain the emission spectrum. We observed that whatever its initial site the sodium atom moves very rapidly to an interstitial site between two planes of the matrix where the emission occurs. This phenomenon should explain the only emission band observed experimentally.
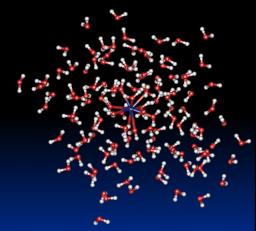
In this field, the main goal relates to the description of physical phenomena involved in the solvation of an ion and we treat this problem performing classical molecular dynamics simulations of confined clusters. Previously, theses studies require the parameterization of model potentials that is done by carrying out sophisticated ab initio calculations on small systems. The model potentials take into account the main interaction energy terms, i.e. an electrostatic term, a polarization term, repulsion and dispersion terms. The main advantages of theses model potentials are on the one hand, that the electrostatic model is built on atomic multipoles up to quadrupoles and on the other hand, that the polarization term is described as a many-body term using a self consistent dipole polarization procedure.