Pages scientifiques 2006

L’activité du groupe est centrée sur l’interaction de lasers ultracourts avec la matière condensée, dans une gamme d’éclairement comprise entre 1012W/cm2 et 1019W/cm2. Les expériences nécessitant un éclairement élevé sont réalisées sur une installation laser mise à notre disposition (LASER UHI10), l’accès au serveur LUCA nous est ouvert pour des campagnes à éclairements plus modestes. Deux thèmes sont abordés :
les propriétés optiques des solides très excités : notre objectif scientifique est de caractériser le comportement de la matière à la densité du solide, dans des régimes d’éclairements modérés, typiquement entre 1012 et 1015W/cm2. Nous étudions principalement le phénomène de claquage induit par laser dans les matériaux ainsi que la transition de l’état de matériau transparent à celui de plasma réfléchissant dans un diélectrique.
l’interaction laser-matière à ultra-haute intensité (1016-1019W/cm2). La course aux éclairements élevés a permis en quelques années d’accéder à un régime d’interaction pour lequel l’énergie conférée aux particules atteint le domaine relativiste. Des résultats récents montrent qu’il est possible d’accélérer des ions à plusieurs dizaines de MeV, d’obtenir des électrons rapides monoénergétiques, de produire des neutrons par réaction de fusion, ou encore de générer des photons X par conversion de fréquence dans le matériau irradié. Dans cette gamme d’éclairement élevé, nous souhaitons caractériser et développer des sources de courte longueur d’onde en vue de leurs applications. En particulier, nous étudions :
- la génération d’harmoniques d’ordre élevé par interaction laser-solide en régime relativiste .
- l’utilisation du rayonnement harmonique généré en gaz pour sonder des plasmas denses et chauds créés par laser .
- la génération de rayonnement X incohérent créé à partir d’irradiation intense de poudres microniques .
- Génération de particules et applications à l’étude de dégâts infligés aux matériaux. Ces études seront menées dans une salle radioprotégée, actuellement en cours d’aménagement.
Les matériaux peuvent, comme les molécules et les agrégats, être excités électroniquement par les photons, les ions et les électrons. Les mesures résolues en temps utilisant des impulsions lasers ultra-brèves et intenses donnent accès à un régime temporel où les mouvements atomiques (vibrations du solide, expansion hydrodynamique du plasma) peuvent être considérés comme gelés. Ces conditions permettent une exploration directe des phénomènes de relaxation électronique et de transfert d'excitation entre les électrons et le réseau (création de défauts en particulier). Ces fortes densités d’excitation sont à rapprocher de celles créées au voisinage des traces d’ions multichargés. Par ailleurs, des états jusqu'alors inexplorés de la matière sont obtenus lorsque celle-ci est soumise quasi-instantanément à des champs électriques de l'ordre de l'unité atomique.
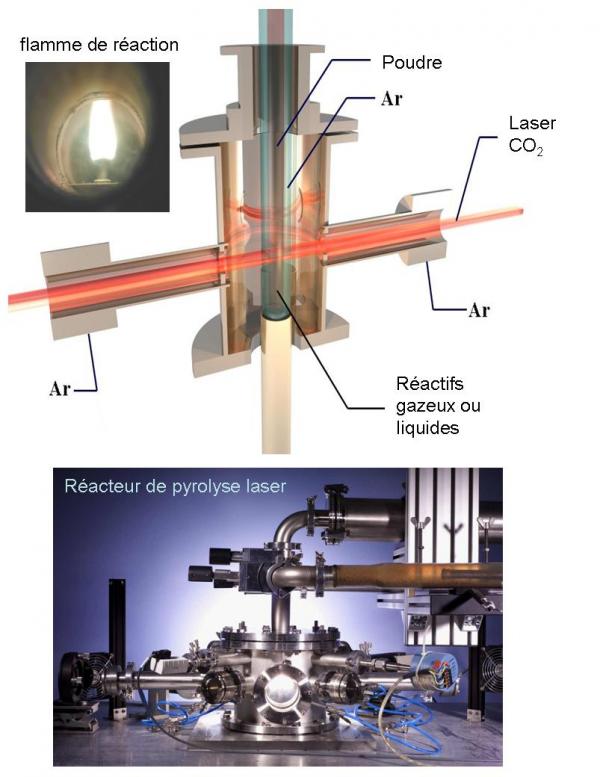
Principe de la pyrolyse laser
Il repose sur l’interaction en jets croisés entre un faisceau laser infrarouge CO2 et un flux de réactifs dans un réacteur sous atmosphère contrôlée. Le transfert d’énergie provoque une élévation de température dans la zone de réaction, les précurseurs sont dissociés, une flamme apparaît dans laquelle des nanoparticules sont formées sans interaction avec les parois du réacteur. Les précurseurs peuvent être gazeux ou liquides. Dans le cas d’un liquide, le précurseur est injecté dans le réacteur sous forme d’aérosol.
Parmi les différentes méthodes de synthèse de nano-objets, la pyrolyse laser se distingue par sa souplesse et la variété des composés qu’elle peut produire tant en termes de composition chimique que de morphologie et de cristallinité.
Les étapes du processus sont :
- Excitation des états vibrationnels des molécules absorbant le rayonnement infrarouge
- Transfert de l’excitation par collision à toutes les molécules du milieu
- Dissociation des molécules donnant lieu à une vapeur saturée
- Nucléation homogène
- Croissance des nanoparticules
Le rendement chimique peut être supérieur à 90% pour les précurseurs gazeux.
Les taux de production sont compris entre 30 et 100 g/h pour les nanopoudres à base de Si en laboratoire et peuvent atteindre 1 kg/h sur pilote de production pour des nanoparticules de SiC.
Les principaux paramètres ajustables sont :
- La nature des précurseurs (par exemple : SiH4, C2H4, HMDS, TEOS, … .)
Grâce à cette grande variété de précurseurs, différentes poudres nanostructurées ont déjà été obtenues par pyrolyse laser, par exemple : Si, SiC, Si/C/N, Si/C/N/Y/Al, Si/C/B/N, Si/C/O, a-C:H, C, C60, C70, C-N, B4C, TiC, WC, FeC, Fe3C, TiB2, ZrB2, Fe4N, Fe, FeO, Fe2O3, TiO2, Al2O3, V2O5, CrO2, etc. - La puissance laser
Elle permet notamment la maîtrise de la température de flamme et donc de la cristallinité des produits. - Le débit des réactifs
Il régit le temps de séjour des réactifs dans la flamme. Il permet ainsi d’ajuster la taille des nanoparticules dans une large gamme.

Les travaux sur le couplage réducteur des cétones par le système MCl4-Li(Hg) (M = Ti, U) ont révélé les différentes voies pouvant être suivies par la réaction de McMurry. La première étape est toujours la formation du radical cétyle (voie a) qui, lorsque l'encombrement stérique le permet, se dimérise en métallapinacol (voie b) pour donner ensuite le diol α ou l'oléfine de couplage (voies c et d). Ce couplage peut être dissymétrique dans le cas des cétones aromatiques encombrées (voie b'). Cependant, lorsque les substituants R sont volumineux (R = iPr), le radical cétyle ne peut se coupler et est alors désoxygéné en métallacarbène (voie e) qui peut ensuite réagir avec la cétone pour fournir l'oléfine de couplage (voie f) ou se réarranger en l'oléfine résultant de la désoxygénation de la cétone (voie g). Enfin, les métallacarbènes issus de cétones très encombrées (R = tBu), sont transformés en alcanes correspondants, par abstraction d'un atome d'hydrogène du solvant ou par hydrolyse (voie h).
Le mécanisme de la réaction de McMurry n'est donc pas limité aux étapes a - d, comme il était généralement admis. En suivant les voies e - h, cette réaction présente des analogies avec les réactions de Wittig et de Clemmensen.
L'étude des propriétés chimiques d'édifices moléculaires en solution ou aux interfaces ne peut éluder la nature quantique des électrons. La dynamique moléculaire classique doit alors faire place à la dynamique moléculaire ab initio (par ex., de type Car-Parrinello). Dans ce cadre, notre champ de recherche comprend en amont le développement de pseudopotentiels associés à une base d'onde planes et en aval le calcul de profils d'énergie libre.
La dynamique moléculaire est mise en échec par les phénomènes de temps long. À l'opposé, les méthodes statistiques pures (théories de l'état de transition ou RRKM) s'affranchissent de ce problème par des évaluations de volume d'espace de phase au prix d'une hypothèse de dynamique intra rapide mal vérifiée. Nous avons mis au point un traitement mixte statistique dynamique qui incorpore la dynamique intra au moyen d'équations cinétiques sur les populations d'états. Ce formalisme peut s'appliquer tant à des réactions chimiques qu'à des processus physicochimiques (évaporation, diffusion par sauts).
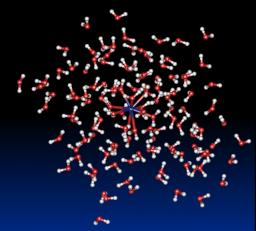
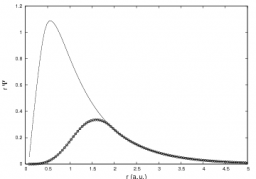
L'objectif de cette thématique est la description des phénomènes physiques impliqués dans la solvatation d’ions étudiée à partir de simulations de dynamique moléculaire classique d'agrégats confinés. Ces études nécessitent au préalable la mise au point de potentiels modèles qui sont dérivés de calculs ab initio de haut niveau sur des systèmes modèles de petite taille. Ils prennent en compte les principaux termes d'énergie d'interaction, c’est-à-dire, un terme électrostatique, un terme de polarisation, un terme de répulsion et un terme de dispersion. Les deux atouts de ces potentiels sont d'une part une description sophistiquée du terme électrostatique décrit comme l'interaction de distribution multipolaire multicentrique allant jusqu'au quadrupole et d'autre part la modélisation du terme de polarisation comme un terme à n-corps qui inclut ce que l'on appelle communément le terme de retour, c’est-à-dire, que l'on effectue un calcul autocohérent du terme de polarisation (polarisation mutuelle des sites polarisables).
Nous avons développé une méthode originale de simulation par dynamique moléculaire de la formation d’une matrice de gaz rare contenant une impureté moléculaire avec calcul des spectres d’absorption et des spectres d’excitation de la molécule piégée dans la matrice.
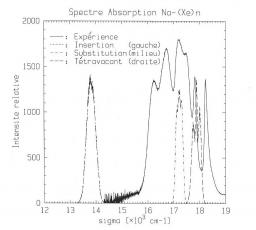
Codéposition de sodium en matrice de xénon : Dans une collaboration théorie-expérience avec C. Crépin (LPPM, Orsay), J. G. McCaffrey et M. Ryan (NUI Maynooth, Irlande) nous avons étudié par dynamique moléculaire (DM) la codéposition d’atomes de sodium en matrice de xénon. On s’est intéressé à la caractérisation, structure et énergie de cohésion, des sites d’occupation les plus probables ainsi qu’au calcul dynamique des spectres d’absorption et d’émission du sodium piégé dans la matrice. Seul le site de substitution où l’atome de sodium ne remplace qu’un atome de xénon a été observé à l’issue des simulations. La comparaison de spectres d’absorption calculés pour différents jeux de potentiels a montré que la méthode était un test sévère de leur précision. Le spectre d’émission est obtenu par une DM réalisée sur l’état excité. On a observé que quel que soit le site originel l’atome de sodium excité se retrouve très rapidement dans un site d’insertion entre deux plans de la matrice à partir duquel a lieu l’émission. Ce phénomène explique la bande unique observée expérimentalement en émission.<