








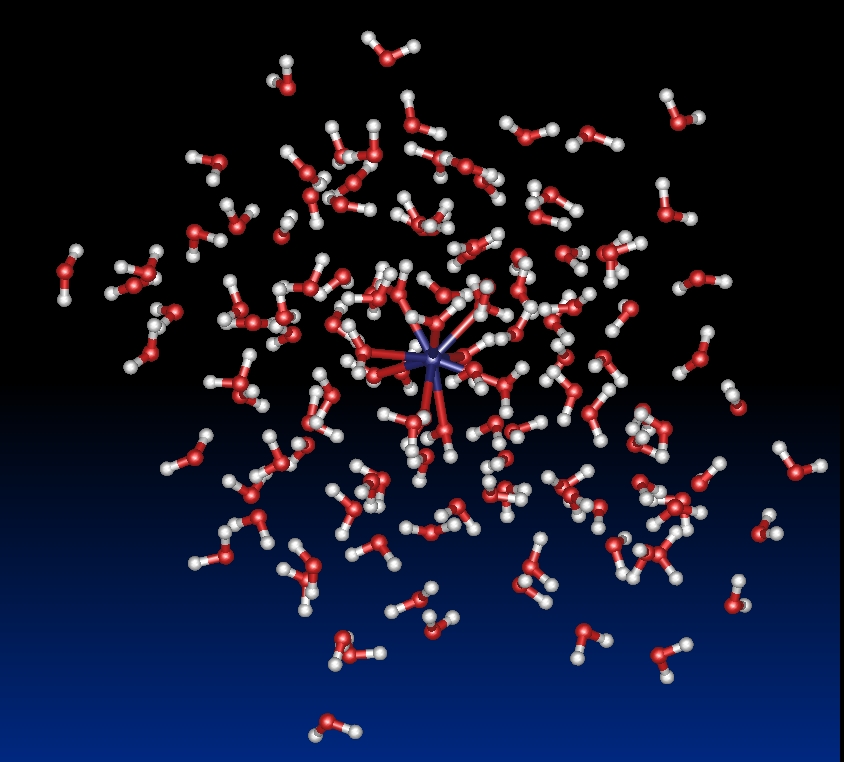
[La-(H20)60]3+ cluster
In this field, the main goal relates to the description of physical phenomena involved in the solvation of an ion and we treat this problem performing classical molecular dynamics simulations of confined clusters. Previously, theses studies require the parameterization of model potentials that is done by carrying out sophisticated ab initio calculations on small systems. The model potentials take into account the main interaction energy terms, i.e. an electrostatic term, a polarization term, repulsion and dispersion terms. The main advantages of theses model potentials are on the one hand, that the electrostatic model is built on atomic multipoles up to quadrupoles and on the other hand, that the polarization term is described as a many-body term using a self consistent dipole polarization procedure.
Hydration of lanthanides (III) ions: We present the results obtained with this new approach based on a molecular dynamics simulation (1 - 2 ns, T=300K) of [Ln-(H2O)n]3+ clusters (Ln3+= La3+, Eu3+ and Gd3+, n = 60 – 200) confined by spherical boundary conditions and taking into account many-body effects. The results show for the first time an excellent agreement with available experimental data for the structures of the first two coordination shells and dynamical properties such as the water exchange rate between the first and second coordination shell. At 4.63 Å for La3+, respectively 4.64 Å for Eu3+ and 4.65 Å for Gd3+, a second coordination shell is clearly identified with about 16 (La3+, Eu3+) - 18 (Gd3+) water molecules. This result is in good agreement with the single available experimental data obtained by Large Angle X-ray Scattering (LAXS) for the La3+ (18 water molecules at 4.63 Å). In agreement with experimental data, an analysis of the molecular dynamics trajectories shows that an arrangement with a coordination number of 9 is the most recurrent for La3+ whereas an equilibrium value between 9 and 8 is the preferred arrangement for Eu3+ and Gd3+. Furthermore, the magnitude of the charge transfer between the gadolinium ion and the solvent was studied thanks to the implementation of a fluctuating charge model. It was established that a significant charge transfer exists, about one electron, and that it is highly correlated to the instantaneous coordination of the ion.
Rafial distribution function gLa-O(r), gLa-H (r) and integration curve for La(III)
Application to the study of contrast agents involved in MRI: The chemical class of gadolinium (III) polyaminocarboxylate complex is currently used as contrast agents in MRI in order to improve the visualization of cancerous tumors. Their efficiency is largely determined by the exchange rate of the water molecules that belong to the first coordination sphere with their environment. A non-empirical approach in order to design new contrast agents implies the development of tools to carry out realistic simulations. The methodology mentioned above was extended to study such complexes. We obtain the first quantitative molecular dynamics simulations able to reproduce experimental data for coordination numbers and residence time of the water molecules in the first coordination sphere, and also to provide new structural data for the first coordination spheres and magnitude of the charge transfer between the ion and the ligand.
Further informations:
Molecular dynamics study of the hydration of lanthanum(III) and Europium(III) including many-body effects C. Clavaguera, R. Pollet, J.M. Soudan, V. Brenner, J.P. Dognon J. Phys. Chem. B 2005, 109, 7614.
Theoretical study of the hydrated Gd3+ ion: structure, dynamics and charge tranfer C. Clavaguera, F. Calvo, J.P. Dognon, J. Chem. Phys. 2006, 124, 074505.
• ![]() Laser-matter interaction › Physical Chemistry Synthesis and characterization of nano-objects › Supramolecular, structural and coordination chemistry
Laser-matter interaction › Physical Chemistry Synthesis and characterization of nano-objects › Supramolecular, structural and coordination chemistry
• Service des Photons Atomes et Molécules • Service des Photons Atomes et Molécules
• Theory
