A new Monte Carlo method for getting the density of states of atomic cluster systems
J. M. Soudan, M. Basire, J. M. Mestdagh and C. Angelie
J. Chem. Phys., 135, 144109, 2011 [doi]
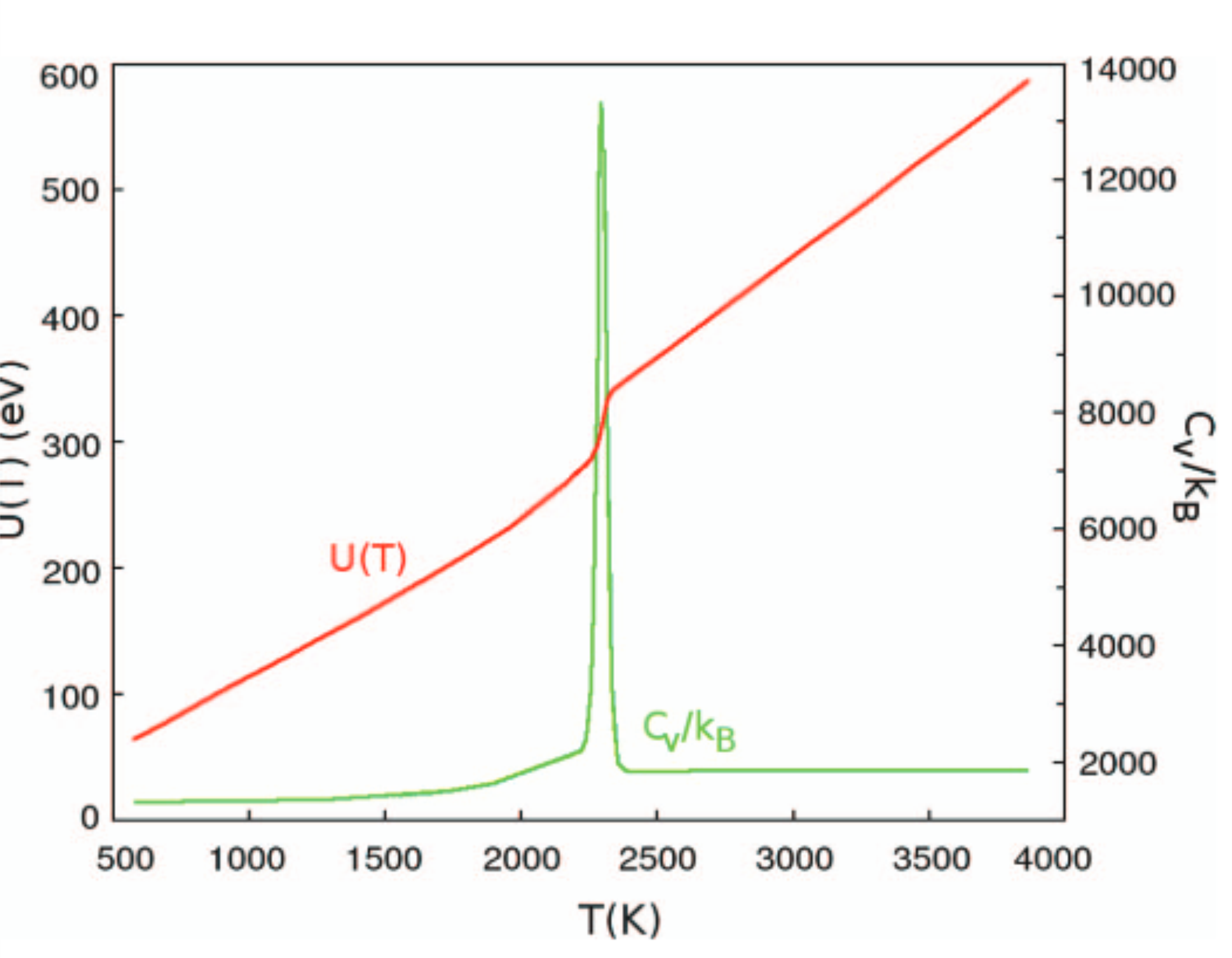
A novel Monte Carlo flat histogram algorithm is proposed to get the classical density of states in terms of the potential energy, g(E-p), for systems with continuous variables such as atomic clusters. It aims at avoiding the long iterative process of the Wang-Landau method and controlling carefully the convergence, but keeping the ability to overcome energy barriers. Our algorithm is based on a preliminary mapping in a series of points (called a sigma-mapping), obtained by a two-parameter local probing of g(E-p), and it converges in only two subsequent reweighting iterations on large intervals. The method is illustrated on the model system of a 432 atom cluster bound by a Rydberg type potential. Convergence properties are first examined in detail, particularly in the phase transition zone. We get g(E-p) varying by a factor 10(3700) over the energy range [0.01 < E-p < 6000 eV], covered by only eight overlapping intervals. Canonical quantities are derived, such as the internal energy U(T) and the heat capacity C-V(T). This reveals the solid to liquid phase transition, lying in our conditions at the triple point. This phase transition is further studied by computing a Lindemann-Berry index, the atomic cluster density n(r), and the pressure, demonstrating the progressive surface melting at this triple point. Some limited results are also given for 1224 and 4044 atom clusters. (C) 2011 American Institute of Physics. [doi: 10.1063/1.3647333]

Nanothermodynamics of large iron clusters by means of a flat histogram Monte Carlo method
M. Basire, J. M. Soudan and C. Angelie
J. Chem. Phys., 141, 104304, 2014 [doi]
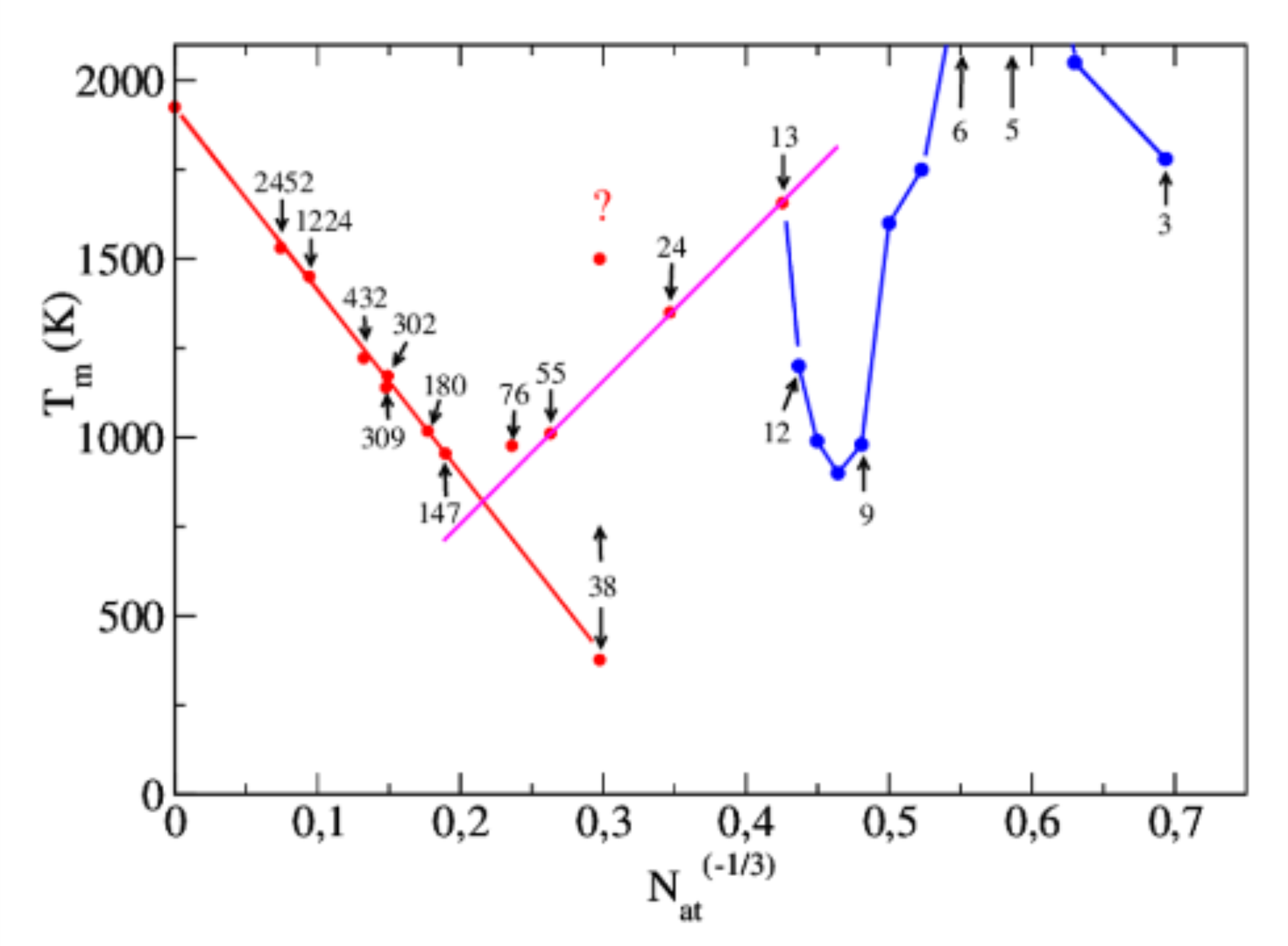
The thermodynamics of iron clusters of various sizes, from 76 to 2452 atoms, typical of the catalyst particles used for carbon nanotubes growth, has been explored by a flat histogram Monte Carlo (MC) algorithm (called the σ-mapping), developed by Soudan et al. [J. Chem. Phys. 135, 144109 (2011), Paper I]. This method provides the classical density of states, gp(Ep) in the configurational space, in terms of the potential energy of the system, with good and well controlled convergence properties, particularly in the melting phase transition zone which is of interest in this work. To describe the system, an iron potential has been implemented, called “corrected EAM” (cEAM), which approximates the MEAM potential of Lee et al. [Phys. Rev. B 64, 184102 (2001)] with an accuracy better than 3 meV/at, and a five times larger computational speed. The main simplification concerns the angular dependence of the potential, with a small impact on accuracy, while the screening coefficients Sij are exactly computed with a fast algorithm. With this potential, ergodic explorations of the clusters can be performed efficiently in a reasonable computing time, at least in the upper half of the solid zone and above. Problems of ergodicity exist in the lower half of the solid zone but routes to overcome them are discussed. The solid-liquid (melting) phase transition temperature Tm is plotted in terms of the cluster atom number Nat. The standard N−1/3at linear dependence (Pawlow law) is observed for Nat >300, allowing an extrapolation up to the bulk metal at 1940 ±50 K. For Nat <150, a strong divergence is observed compared to the Pawlow law. The melting transition, which begins at the surface, is stated by a Lindemann-Berry index and an atomic density analysis. Several new features are obtained for the thermodynamics of cEAM clusters, compared to the Rydberg pair potential clusters studied in Paper I
Nanothermodynamics of iron clusters: Small clusters, icosahedral and fcc-cuboctahedral structures
C. Angelie and J. M. Soudan
J. Chem. Phys., 146, 174303, 2017 [doi]
The study of the thermodynamics and structures of iron clusters has been carried on, focusing on small clusters and initial icosahedral and fcc-cuboctahedral structures. Two combined tools are used. First, energy intervals are explored by the Monte Carlo algorithm, called sigma-mapping, detailed in the work of Soudan et al. [J. Chem. Phys. 135, 144109 (2011), Paper I]. In its flat histogram version, it provides the classical density of states, g(p)(E-p), in terms of the potential energy of the system. Second, the iron system is described by a potential which is called "corrected EAM" (cEAM), explained in the work of Basire et al. [J. Chem. Phys. 141, 104304 (2014), Paper II]. Small clusters from 3 to 12 atoms in their ground state have been compared first with published Density Functional Theory (DFT) calculations, giving a complete agreement of geometries. The series of 13, 55, 147, and 309 atom icosahedrons is shown to be the most stable form for the cEAM potential. However, the 147 atom cluster has a special behaviour, since decreasing the energy from the liquid zone leads to the irreversible trapping of the cluster in a reproducible amorphous state, 7.38 eV higher in energy than the icosahedron. This behaviour is not observed at the higher size of 309 atoms. The heat capacity of the 55, 147, and 309 atom clusters revealed a pronounced peak in the solid zone, related to a solid-solid transition, prior to the melting peak. The corresponding series of 13, 55, and 147 atom cuboctahedrons has been compared, underscoring the unstability towards the icosahedral structure. This unstability occurs clearly in several steps for the 147 atom cluster, with a sudden transformation at a transition state. This illustrates the concerted icosahedron-cuboctahedron transformation of Buckminster Fuller-Mackay, which is calculated for the cEAM potential. Two other clusters of initial fcc structures with 24 and 38 atoms have been studied, as well as a 302 atom cluster. Each one relaxes towards a more stable structure without regularity. The 38 atom cluster exhibits a nearly glassy relaxation, through a cascade of six metastable states of long life. This behaviour, as that of the 147 atom cluster towards the amorphous state, shows that difficulties to reach ergodicity in the lower half of the solid zone are related to particular features of the potential energy landscape, and not necessarily to a too large size of the system. Comparisons of the cEAM iron system with published results about Lennard-Jones systems and DFT calculations are made. The results of the previous clusters have been combined with that of Paper II to plot the cohesive energy E-c and the melting temperature T-m in terms of the cluster atom number N-at. The N-at(-1/3) at linear dependence of the melting temperature (Pawlow law) is observed again for N-at > 150. In contrast, for N-at < 150, the curve diverges strongly from the Pawlow law, giving it an overall V-shape, with a linear increase of T-m when N-at goes from 55 to 13 atoms. Surprisingly, the 38 atom cluster is anomalously below the overall curve. Published by AIP Publishing.