


Liquide et solide désordonné se distinguent en premier lieu par leur viscosité : l’un coule, l’autre pas. Mais existe-t-il de réelles différences de structure entre ces deux états de la matière et quelle est la nature exacte la transition « vitreuse » qui les sépare ? Si des réponses ont pu être apportées pour des liquides composés d’atomes ou de petites molécules, ces questions se posent aussi pour les polymères fondus, constitués de longues chaines de molécules. Cette question est technologiquement d’importance, puisque la solidification de liquides polymères est au cœur de beaucoup de procédés, tel que le façonnage de la matière plastique.
Une collaboration de spécialistes du domaine, incluant des chercheurs du LLB, montre toute la spécificité du problème de la transition vitreuse pour les polymères, qui font apparaitre une dimension caractéristique supplémentaire : leur longueur de chaine. Si pour les courtes longueurs de chaine, un comportement générique est bien observé à la température de transition vitreuse, des déviations significatives apparaissent pour les longues chaines. Il est alors montré que l’accord peut être rétabli en considérant une propriété dynamique plus macroscopique, telle que la viscosité du polymère.

Les polymères sont de très grosses molécules composées de milliers, voire de millions d’atomes, formant de longues chaines. Ils sont les constituants d’un grand nombre d’objets qui nous entourent, tels les plastiques, peintures et colles… et se retrouvent dans les fibres naturelles animales ou végétales. Comprendre ce qui donne aux polymères leurs propriétés uniques est utile pour permettre le développement de nouveaux matériaux fonctionnels pour diverses technologies actuelles et futures. Le façonnage des solides polymères passent souvent par leur fusion et moulage, et les propriétés finales sont fonction de la façon dont se produit la solidification du liquide initial.
En mécanique, un comportement fragile indique que le système n’a pas le temps de se relaxer, pendant le temps de la sollicitation ou de l’observation, et se casse donc « brutalement » sous l’effet d’un choc. Un comportement fragile correspond donc à des énergies d’activation effectives élevées pour les phénomènes de diffusion interne au système. Un indice dénommé « indice de fragilité » (m) [*] d’un liquide surfondu permet d’évaluer ce paramètre, à la température de transition vitreuse Tg : il mesure de combien varie le temps de relaxation pour un incrément faible de température près de Tg : quand sa valeur est grande, le liquide est dit « fragile », par opposition à « fort » dans le cas de systèmes comme la silice pour lesquels les propriétés dynamiques sont faiblement affectées par des variations de T.
Une différence notable entre polymères et petites molécules est que cet indice m, au voisinage de la transition vitreuse, peut être de 1.5 à 2 fois plus élevé pour les polymères à grande longueur de chaine, première indication d’un comportement très particulier à Tg. Jusqu’à présent, il n’y avait pas de réponse claire pour expliquer cette valeur particulièrement élevée et la différence de comportement entre polymères courts et longs. En combinant plusieurs techniques d’études, dont la diffusion de neutrons, la collaboration de chercheurs issus des États-Unis, d’Italie et de Chine, avec des chercheurs du LLB à Saclay, a pu obtenir une image plus complète du phénomène dans les polymères et préciser comment les polymères fondus diffèrent des liquides de petites molécules. Leur analyse est publiée dans The Journal of Chemical Physics, de AIP Publishing.
[*] L’indice de fragilité m est un indice « réduit » sans dimension. Il est défini comme le rapport de l’énergie d’activation apparente mesurée à Tg, sur la température de transition vitreuse : E(Tg)/Tg.

Le polymère étudié est un polymère modèle : le polystyrène. En comparant plusieurs polymères avec différentes longueurs de chaîne (avec un poids moléculaire MW entre 580 et 223 000), les chercheurs ont dans un premier temps mesuré un « indice de fragilité » compris entre m = 68 pour les chaines courtes (MW = 580), à m=143 pour MW = 223 000. Ils montrent ensuite que les relations usuellement observées entre cet indice de la dynamique m et diverses grandeurs caractéristiques du matériau, telles que le rapport entre le module d’élasticité isostatique et le module de cisaillement, ou l’amplitude du processus de relaxation, restent valables pour les polymères courts, mais ne sont plus vérifiées pour les grandes longueurs de chaine. Ils sont ensuite parvenus pour la première fois à montrer que ces relations peuvent être rétablies si l’on considère un indice de fragilité mchain déduit des mesures de viscosité, au lieu du paramètre usuel m, directement déduit des mesures de temps de relaxation par spectroscopie diélectrique par exemple..
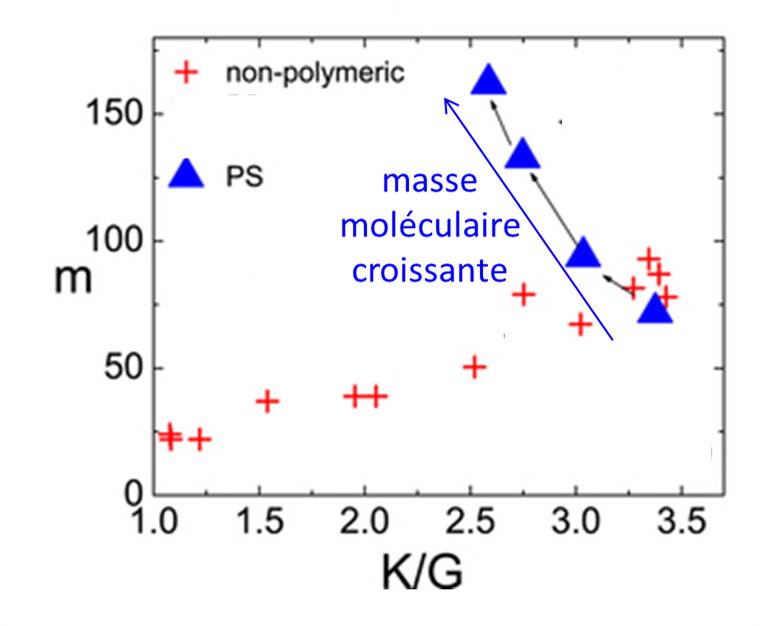
Relation entre « l’indice de fragilité » m et le rapport du module d’élasticité isostatique au module de cisaillement K/G. Pour les polymères courts ou les liquides moléculaires, le paramètre m est simplement proportionnel à ce paramètre mécanique. Une valeur similaire est obtenue pour le polystyrène (PS) de faible masse et une forte déviation est clairement observée pour le PS de forte masse, i.e. à longue chaine polymérique.
Pourquoi cet indice mchain est-il alors une meilleure grandeur pour caractériser les polymères à longue chaine au voisinage de leur transition vitreuse ? Une réponse est apportée en considérant la relation établie par Adam et Gibbs en 1965 [3] entre dynamique et thermodynamique, entre temps de relaxation ou viscosité et entropie configurationnelle à une température donnée. Or pour les polymères de haut poids moléculaires on constate qu’une loi purement exponentielle sur toute la gamme de température explorée jusqu’à Tg est obtenue pour la viscosité, tandis qu’à l’approche de Tg, le temps de relaxation τ dévie de pratiquement un ordre de grandeur. Si toutes les configurations possibles des chaines de polymères sont à prendre en compte pour décrire la viscosité, il est montré que l’augmentation du temps de relaxation est liée à une réduction d’entropie du fait de la rigidité des chaines les plus longues, qui interdisent certaines configurations.
Les polymères à longue chaine rejoignent ainsi une description unifiée du phénomène de transition vitreuse, à condition de bien prendre en compte leur fragilité mchain déduite de l’évolution de leur viscosité, plutôt que celle (m) déduite du temps de relaxation des chaines moléculaires. Ce résultat indique de façon forte que le comportement du polymère fondu est dominé par les phénomènes de relaxation locale qui pilotent la viscosité, plutôt que par le comportement global des chaines, qui induisent des temps de relaxation élevés. Des différences semblables sont à considérer selon la rigidité des chaines de polymères : la forte rigidité d’un polymère entrainant une importante réduction d’entropie et donc un indice de fragilité élevé.
Références :
[1] D’après Prof. J. Lecomte –Beckers, Métallurgie & Science des Matériaux, LTAS – Département d’Aérospatiale et Mécanique, Faculté des sciences appliquées, Université de Liège
[2] Why many polymers are so fragile: A new perspective
C. Dalle-Ferrier, A. Kisliuk, L. Hong, G. Carini Jr., G. Carini, G. D’Angelo, C. Alba-Simionesco, V. N. Novikov, and A. P. Sokolov, The Journal of Chemical Physics 145, 154901 (2016) – http://dx.doi.org/10.1063/1.4964362.
[3] On the temperature dependence of cooperative relaxation properties in glass‐forming liquids
G. Adam and J. H. Gibbs, J. Chem. Phys. 43, 139 (1965). https://doi.org/10.1063/1.1696442
Contact CEA :
Collaboration :
- Laboratoire Léon Brillouin, UMR 12, CEA-CNRS, 91191 Saclay, France
- Chemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
- Institute of Natural Sciences & Department of Physics and Astronomy, Shanghai Jiao Tong University, Shanghai 200240, China
- IPCF del CNR, UOS di Messina, I-98158 Messina, Italy
- Dipartimento di Fisica e Scienze della Terra, Università di Messina, I-98166 Messina, Italy
- Department of Chemistry and Joint Institute for Neutron Sciences, University of Tennessee, Knoxville, Tennessee 37996, USA
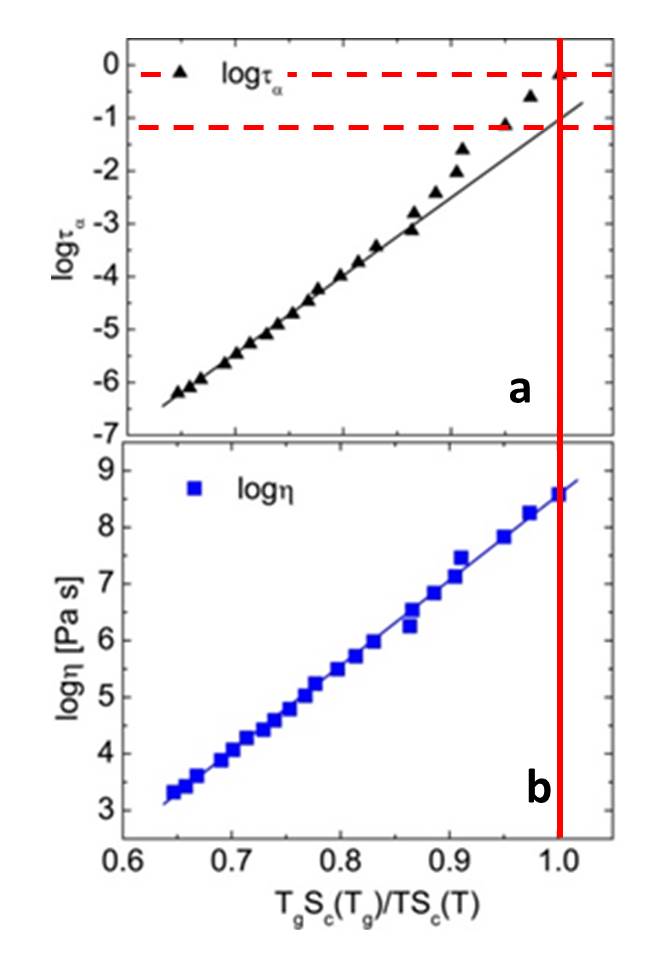
Relations d’Adam-Gibbs (1965) entre le temps de relaxation τ ou la viscosité η et le paramètre entropique 1/TSc(T) (en coordonnées réduites à Tg). Sc désigne l’entropie configurationnelle, déduite de mesures calorimétriques. a) une déviation d’un ordre de grandeur est observée près de Tg pour le temps de relaxation local τ. Cette déviation est liée à la réduction d’entropie configurationnelle pour les longues chaines de polymères. b) la relation exponentielle attendue est bien conservée jusqu’à Tg si l’on considère la viscosité η de ce même polymère.