L'état électronique d'une molécule réagit très rapidement – à l'échelle de la femtoseconde (10-15 s), voire de l'attoseconde (10-18 s) – à toute perturbation telle qu'une excitation laser, une vibration qui modifie la position relative des noyaux atomiques qui la constitue, ou encore au cours d'une réaction chimique. Suivre en temps réel l'évolution des orbitales électroniques demande ainsi des techniques d'observation permettant d'atteindre cette résolution temporelle attoseconde.
Une telle sonde ultra-rapide est fournie par la réponse non linéaire de la molécule à un champ laser intense. Dans cette réponse, l’électron de valence peut être extrait de la molécule (avec une certaine probabilité d'ionisation), accéléré et « renvoyé » vers la molécule ionisée (recollision), où il peut se désexciter (recombinaison vers l’orbitale de valence), en émettant une impulsion intense de lumière UV attoseconde. De l’analyse complète de cette émission lumineuse (mesure de l'amplitude, phase et polarisation), il est possible de reconstruire le paquet d’ondes électronique dans les orbitales de valence avec une résolution spatiale subnanométrique et de suivre leur dynamique avec une résolution temporelle attoseconde.
Le travail publié dans les références [1,2,3,4] est le fruit de la collaboration entre le groupe Attophysique du SPAM, qui a réalisé les études expérimentales sur le laser LUCA de l’IRAMIS, et le groupe théorique du Laboratoire de Chimie Physique-Matière et Rayonnement de l’Université P&M Curie. Les études, centrées sur la molécule N2, montrent comment l’émission attoseconde peut caractériser la dynamique des orbitales électroniques induite dans des petites molécules par un champ (laser) intense.
Suivre en temps réel l'évolution – à l'échelle femto ou attoseconde (1 as = 10-18 s)- des orbitales électroniques sous l'effet d'une perturbation, n'est pas si simple. Le principe de la méthode utilisée est le suivant : soumis à l'excitation d'une impulsion laser brève et intense, la barrière tunnel d'un électron entre son état lié et l'état libre est abaissée et le système moléculaire peut être ionisé par cette voie. L'électron éjecté est ensuite rétro-accéléré par le champ électrique intense de l'impulsion laser qui oscille à la fréquence de l'onde lumineuse (période de l'ordre de la femtoseconde : ~ 10-15 s) et vient ainsi « recollisionner » à cette fréquence l'ion formé. La recombinaison radiative possible à chaque recollision, vers les états libres de la molécule, conduit à la génération d’un train d'impulsions de ~ 100 attosecondes = 10-16 s, correspondant à une grande largeur spectrale dans l’extrême-UV (1-100 nm). Le spectre associé à cet ensemble d'impulsions montre la génération d'harmoniques d’ordres élevés de l'impulsion laser initiale.
Dans le cas étudié de la molécule N2, le problème est un peu plus complexe puisque le processus est fonction de l'angle θ entre l'axe de symétrie de la molécule (défini par les deux noyaux) et le champ électrique de l'impulsion laser (polarisation). Les molécules sont alors préalablement alignées dans le référentiel du laboratoire par une première impulsion laser femtoseconde (alignement impulsionnel).
La caractérisation complète de l’émission lumineuse (amplitude et phase des deux composantes de polarisation, pour tous les ordres harmoniques – méthode RABBIT), donne accès au moment dipolaire électrique associé à la recombinaison radiative entre les orbitales « libre » (ion) et liée (états de valence de la molécule). A partir du dipôle ainsi déterminé, il est ensuite possible de reconstruire une image (amplitude et phase) des orbitales impliquées dans les transitions, avec une résolution de l’ordre du dixième de nanomètre (fraction de la longueur d’onde de de Broglie associée à l’électron incident) et de suivre leur dynamique avec une résolution temporelle de quelques 100 as (la durée de la recollision) à quelques femtosecondes (la durée du train des recollisions successives [5, 6].
Ce dipôle moléculaire (amplitude et phase dans un grand domaine spectral) contient une information très riche sur la dynamique électronique et nucléaire dans la molécule. Ceci est illustré par deux séries d’études :
– Dans une première série d’études [2,3], le dipôle moléculaire révèle la contribution de plusieurs orbitales de valence à l’ionisation par effet tunnel – orbitales HOMO (Highest Occupied Molecular Orbital, 3Σg Eliaison = 15,58 eV) et HOMO-1 (immédiatement inférieure en énergie, A 1Π+u Eliaison = 16,9 eV), la contribution de chacune étant fonction de la puissance laser et de l’alignement de la molécule (angle θ). Les variations associées de l’émission attoseconde permettent de résoudre les contributions des deux orbitales. En particulier, la variation rapide de la phase du dipôle (« saut de phase » de 0.8 π au voisinage des ordres 23-27 – voir Figure 2/droite-(b)) indique que la contribution de l’orbitale HOMO-1 devient dominante à forte intensité laser pour θ=90°, en accord avec les propriétés de symétrie des orbitales dans le champ laser (Figure 2). De plus, une analyse approfondie révèle que la « remontée » de la phase du dipôle pour les ordres > 27 est une signature de la dynamique nucléaire ultrarapide lors du processus d'émission impliquant HOMO-1.
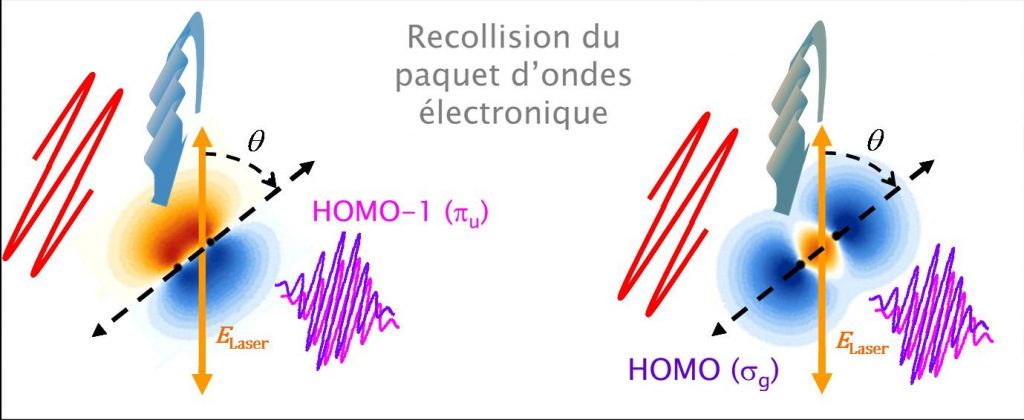
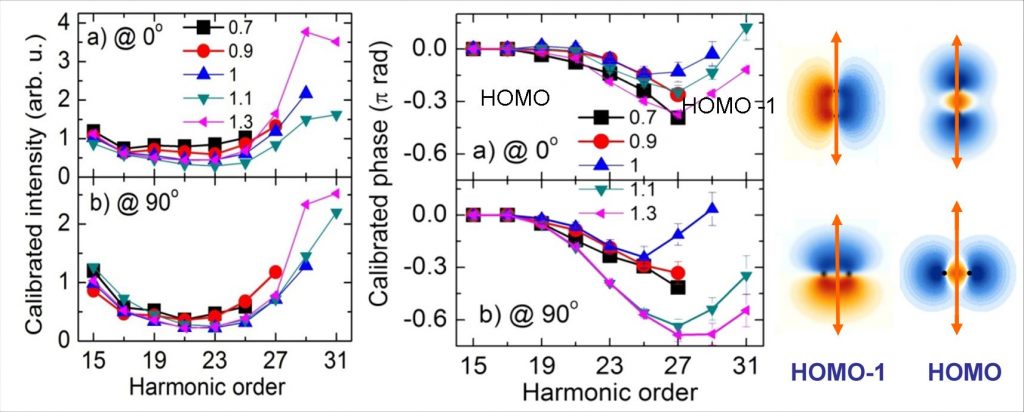
– Dans une seconde série d'études [1,4,6], la mesure du dipôle moléculaire permet de reconstruire « l'image 2D » (en amplitude et en phase) des orbitales impliquées dans l’ionisation tunnel, avec une résolution spatiale de l’ordre du dixième de nanomètre (imposée par la longueur d’onde de de Broglie associée à l’électron incident, voir fait marquant : Imagerie attoseconde d’orbitales moléculaires). On accède également à l’évolution du paquet d’ondes électronique oscillant, créé dans l’ion N2+ par la superposition cohérente des deux voies quantiques d'ionisation HOMO et HOMO-1, avec une résolution temporelle de quelques 100 as (imposée par l'intervalle entre les recollisions) [5, 6].
Ainsi l’émission attoseconde d’une molécule en champ laser intense, et plus généralement le paradigme du « self-probing » par la recollision suivant l'ionisation tunnel, prouvent ici toute leur pertinence comme sonde ultrarapide de la dynamique électronique et nucléaire de la molécule.
Références :
[1] Self-probing of molecules with high-harmonic generation,
S. Haessler, J. Caillat and P. Salières, J. Phys. B 44 (2011) 203001.
[2] Spectrally resolved multi-channel contributions to the harmonic emission in N2,
Z Diveki, A Camper, S Haessler, T Auguste, T Ruchon, B Carré, P Salières, R Guichard, J Caillat, A Maquet, and R Taïeb, New Journal of Physics 14 (2012) 023062.
[3] Molecular Orbital Tomography from Multi-Channel Harmonic Emission in N2, Z. Diveki, R. Guichard, J. Caillat, A. Camper, S. Haessler, T. Auguste, T. Ruchon, B. Carré, A. Maquet, R. Taïeb and P. Salières, Chemical Physics (2012).
[4] Imaging orbitals with attosecond and Angström resolutions: toward attosecond chemistry?,
P. Salières, A. Maquet, S. Haessler, J. Caillat and R. Taïeb, Rep. Prog. Phys. 75 (2012) 062401.
[5] Tomographic imaging of molecular orbitals,
J. Itatani, J. Levesque, D. Zeidler, H. Niikura, H. Pépin, J. C. Kieffer, P. B. Corkum & D. M. Villeneuve, Nature 432 (2004) 867.
[6] Attosecond imaging of molecular electronic wavepackets
S. Haessler, J. Caillat, W. Boutu, C. Giovanetti-Teixeira, T. Ruchon, T. Auguste, Z. Diveki, P. Breger, A. Maquet, B. Carré, R. Taïeb & P. Salières, Nature Physics, 6 (2010) 200.
Contacts :
Z. Diveki1, R. Guichard2, A. Camper1, J. Caillat2, S. Haessler3, T. Ruchon1, T. Auguste1, P. Breger1, A. Maquet2, B. Carré1, R. Taieb2, P. Salières1,
- 1 CEA-Saclay, IRAMIS, Service des Photons, Atomes et Molécules, 91191 Gif-sur-Yvette
- 2 Laboratoire de Chimie Physique-Matière et Rayonnement – LCPMR, CNRS – UPMC, UMR 7614, 11 rue Pierre et Marie Curie, 75231 Paris Cedex 05.
- 3 Photonics Institute, Vienna Univ. of Technology, Gusshausstrasse 27/387, 1040, Vienna, Austria
Contacts CEA : Pascal Salières, Bertrand Carré.