







Theoretical investigations of the electronic states of NaXe: A comparative study
F. Ben Salem, M. B. H. Rhouma, F. Spiegelman, J. M. Mestdagh and M. Hochlaf
J. Chem. Phys., 137, 224310, 2012 [doi]
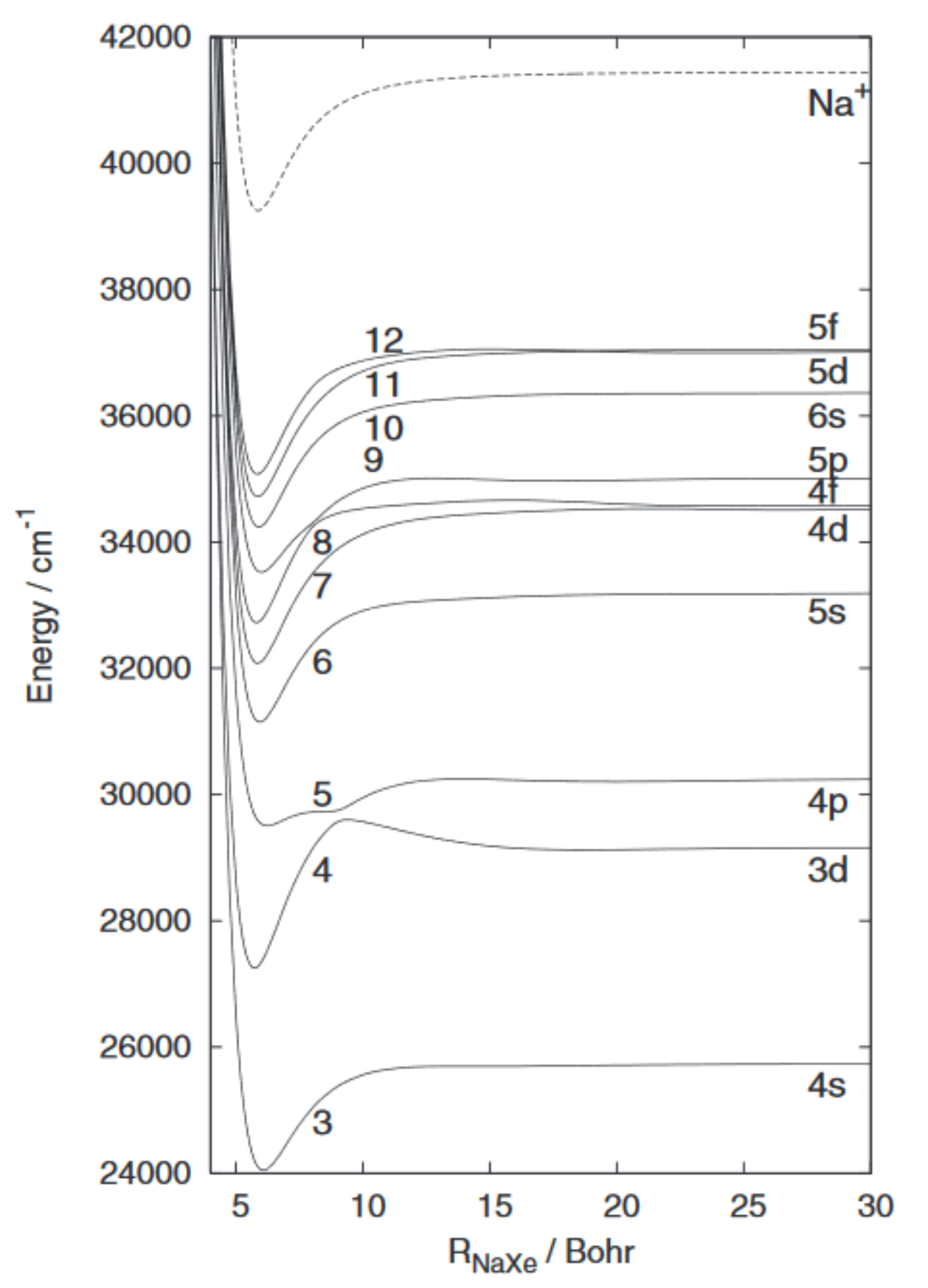
The electronic state properties of NaXe are investigated using ab initio methodologies and various pseudopotential approaches for comparison. The spectroscopic terms and dipole moments of the lowest electronic states up to the Na(3d) +Xe dissociation limit are determined. The difference between valence or smaller core pseudopotential on Xe is shown to be negligible and so is the difference between all-electron and valence pseudopotential completed by core-polarization treatments of Na. These calculations are used as references to test the performance of a treatment involving a zero electron pseudopotential description of xenon together with a one-electron pseudopotential description of Na. When compared with the reference calculations, the one-electron model leads to reasonable quantitative results. The potential energy curves and spectroscopic data of all Rydberg excited states of NaXe up the Na(5f)+Xe dissociation limit are determined using this method. Long distance wells and barriers in the range R = 15-40 bohrs are identified for some of the higher states with (2)Sigma + symmetry. (C) 2012 American Institute of Physics. [http://dx.doi.org/10.1063/1.4769286]
Reactive and Inelastic Channels in the Ca*···FCH3 Transition State: A Simple Branching Mechanism
M. Briant, E. Gloaguen, A. Beswick, J. M. Mestdagh, S. Stolte, L. Poisson, C. Pothier and B. Soep
J. Phys. Chem. A, 2015 [doi]
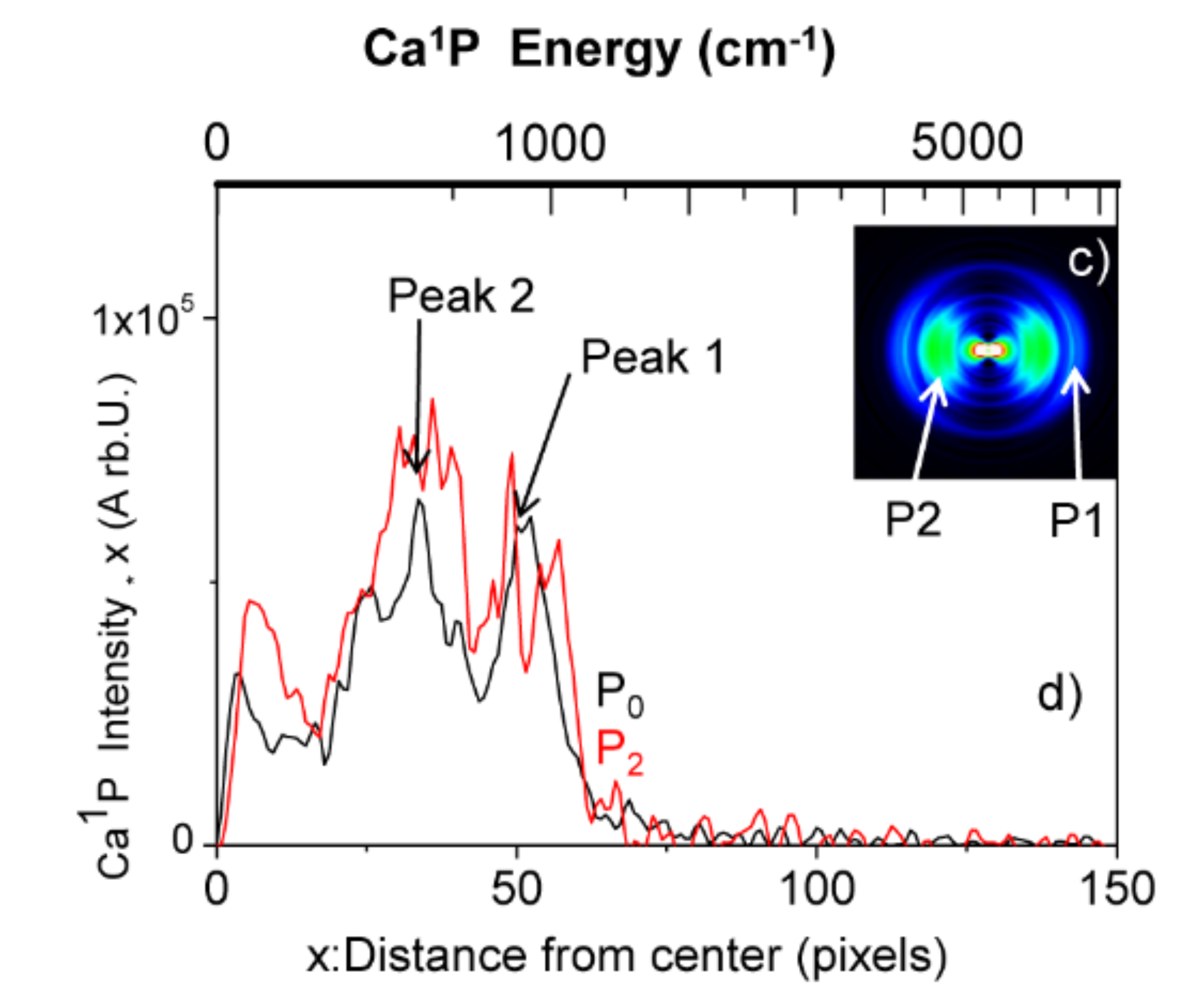
To study the excited state dynamics between a calcium atom and the CH3F molecule, a Ca center dot center dot center dot CH3F 1:1 complex has been prepared by a supersonic expansion with laser ablation of calcium metal in the gas phase. Tunable laser excitation of these complexes in molecular states correlating to Ca P-1(1)(4s4p) + CH3F allows observing two competitive channels: the direct dissociation and the reactive channel into CaF* + CH3. The translational recoil, as well as the alignment of the fragments Ca* and CaF* have been analyzed by velocity map imaging and time-of-flight mass spectrometry. This revealed that both the dissociation and reaction processes are quasi direct and are of comparable intensity. We provide a simple interpretation for this process: the electronically excited potential surface of the Ca*center dot center dot center dot FCH3 complex initiates a fast predissociation from a suspended well to two repulsive surfaces that lead either to Ca P-1(1)(4s4p) (Omega = 1) + CH3F or to CaF((2)Delta) + CH3.
Absorption Spectroscopy, a Tool for Probing Local Structures and the Onset of Large-Amplitude Motions in Small KAr(n) Clusters at Increasing Temperatures
S. Awali, L. Poisson, M. B. E. H. Rhouma and J.-M. Mestdagh
The journal of physical chemistry. A, 119, 9729, 2015 [doi]
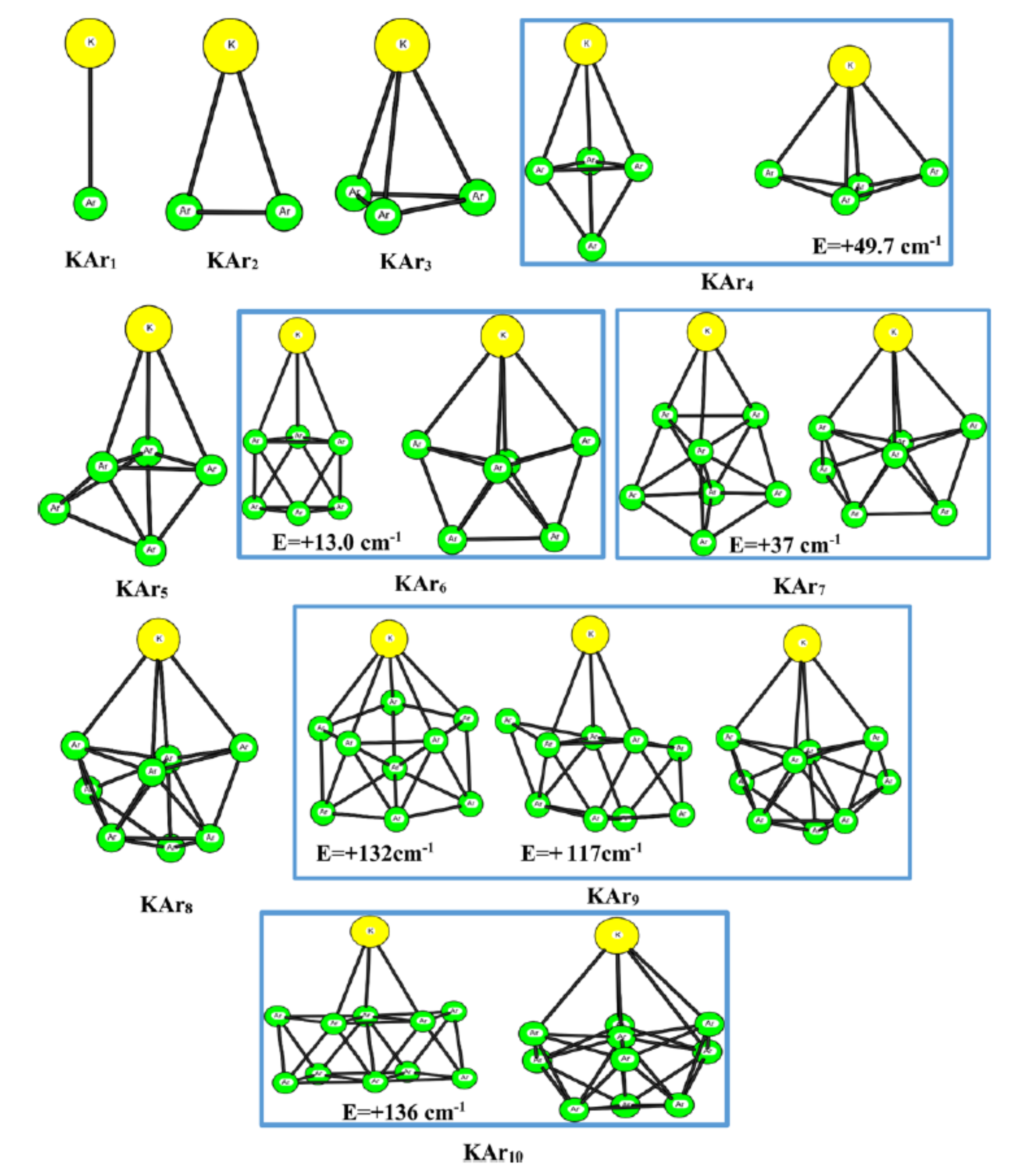
Photoabsorption spectra of KArn (n = 1-10) are simulated at temperatures ranging between 5 and 25 K. The calculations associate a Monte Carlo (MC) method to sample cluster geometries at temperature T, with a one-electron ab initio model to calculate the ground-state and excited-state energies of the cluster. The latter model replaces the K(+) core electrons and all the electrons of the Ar atoms by appropriate pseudopotentials, complemented by core polarization potentials. It also provides the necessary oscillator strengths to simulate the spectra. Global optimization by basin-hopping is used in combination with MC simulation at low temperature (5 K) to identify the most stable isomer and remarkable isomers of ground-state KArn clusters, which are stable with respect to deformations of the order of those expected with Zero Point Energy motions. The absorption spectra calculated for each of these isomers at 5 K suggest that absorption spectroscopy can probe sensitively the local environment of K atom: surface location of K with respect to a close-packed Ar moiety, number of Ar atom in close vicinity, and local symmetry about K. Simulation at increasing temperatures, up to the evaporation limit of K out of the cluster, shows the onset of large amplitude motions above 20 K, when the K atom experiences a variety of local environments.
Large amplitude motion of the acetylene molecule within acetylene-neon complexes hosted in helium droplets
M. Briant, E. Mengesha, P. de Pujo, M. A. Gaveau, B. Soep, J. M. Mestdagh and L. Poisson
Phys. Chem. Chem. Phys., 18, 16414, 2016 [doi]
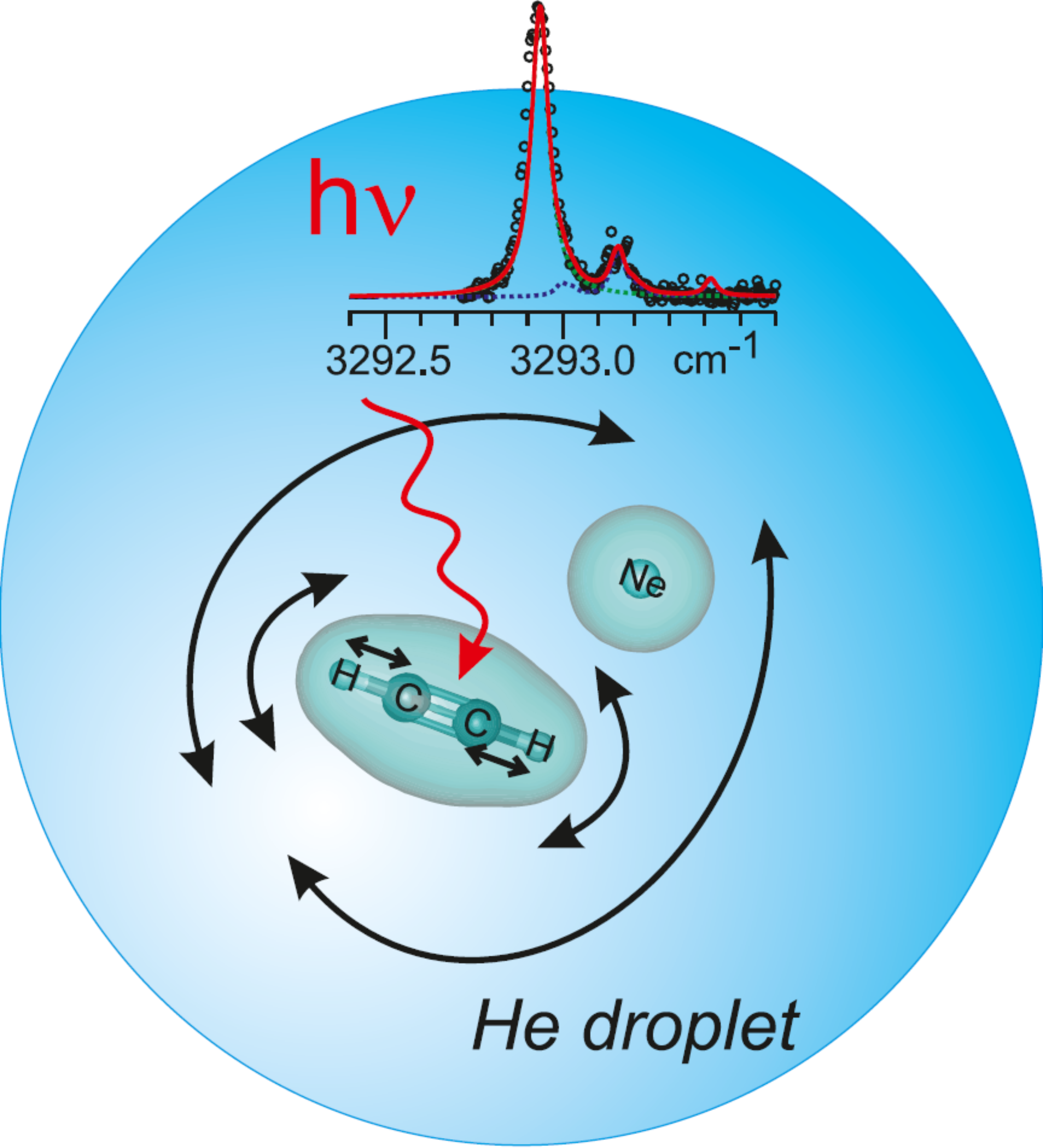
Superfluid helium droplets provide an ideal environment for spectroscopic studies with rotational resolution. Nevertheless, the molecular rotation is hindered because the embedded molecules are surrounded by a non-superfluid component. The present work explores the dynamical role of this component in the hindered rotation of C2H2 within the C2H2-Ne complex. A HENDI experiment was built and near-infrared spectroscopy of C2H2-Ne and C2H2 was performed in the spectral region overlapping the v(3)/v(2) + v(4) + v(5) Fermi-type resonance of C2H2. The comparison between measured and simulated spectra helped to address the above issue.
Dynamics of acetylene dimers hosted in helium droplets
M. Briant, E. Mengesha, M. A. Gaveau, B. Soep, J. M. Mestdagh and L. Poisson
Phys. Chem. Chem. Phys., 20, 2597, 2018 [doi]
The CH antisymmetric stretch of the C2H2 moieties in acetylene dimers was explored over the range 3270-3290 cm-1 using the helium nanodroplet isolation (HENDI) technique. This work is part of a general investigation which addresses the dynamical consequences of coupling the deformation motions of weakly bound complexes with a finite size quantum liquid (the helium droplet). The acetylene dimer is attractive from this point of view because one of its deformation coordinates promotes a tunneling isomerization process. A numerical simulation of the observed spectrum allows deriving a set of effective spectroscopic constants which help understanding the dynamical role played by the droplet on the rotation and deformation of the dimer.

• ![]() Institut Rayonnement Matière de Saclay • Interactions, Dynamics and Lasers Laboratory (LIDYL) - CEA-CNRS and Paris Saclay University
Institut Rayonnement Matière de Saclay • Interactions, Dynamics and Lasers Laboratory (LIDYL) - CEA-CNRS and Paris Saclay University
