









Suite à l'absorption UV, de nombreux systèmes biomoléculaires sont dotés de mécanismes de désactivation des états excités qui assurent leur stabilité photochimique. L'un des principaux objectifs de notre recherche est d'étudier la dynamique conformère-sélective de systèmes moléculaires bio-pertinents par une approche duale expérience-théorie qui consiste en la mise en œuvre :
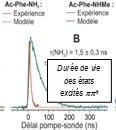
i) d’une caractérisation expérimentale des durées de vie et de la nature des états électroniques formés dans des expériences pompe-sonde nano, pico- et femtoseconde. Des techniques de diagnostic sophistiquées sont utilisées pour caractériser la dynamique d'état excité.
ii) d’une stratégie de calcul multiniveaux multi-étapes innovante afin de caractériser les premiers états excités et de modéliser efficacement leurs surfaces d'énergie potentielle. On utilise en premier lieu des simulations de dynamique non-adiabatique utilisant la théorie de la fonctionnelle de la densité dépendante du temps TDDFT (NA-TDDFT), pour identifier les coordonnées pertinentes. Dans un deuxième temps, des surfaces plus précises sont obtenues par deux types de méthodes à fonction d’onde plus sophistiquées; la méthode « Coupled Cluster » à l’ordre 2 (CC2) et des méthodes multiréférence (CASPT2 et MRCI). Développée sur des modèles de protéines, de petits peptides protégés, cette approche est maintenant appliquée à des peptides protégés microsolvatés ainsi qu'à des dipeptides protégés de chaines latérales diverses. Cela nous a notamment permis de documenter les phénomènes physiques élémentaires contrôlant la durée de vie des états excités de tels systèmes, mettant ainsi en évidence le lien entre la dynamique électronique et la structure. Par ailleurs, un benchmark de la méthode CC2 sur un ensemble de chaînes peptidiques modèles (structures, énergies et fréquences vibrationnelles NH de l’état excité ΠΠ*) ainsi que l'évaluation de la validité de la méthode CC2 par rapport aux méthodes multiréférence ont été réalisés.
Ces travaux s’effectuent dans le cadre du projet ANR ESBODYR (2014-2018) et bénéficient d'un accès aux ressources nationales de calcul haute performance (GENCI) ainsi qu’au serveur SLIC femtoseconde d'IRAMIS (Saclay). Ce projet interdisciplinaire rend ainsi possible les progrès dans la compréhension de la dynamique de l'état excité après absorption de la lumière, ainsi que les développements techniques dans la modélisation du devenir de l'énergie absorbée. Ces processus de conversion d'énergie, en effet, sont également d'une importance primordiale avec des applications potentielles dans de nombreux domaines aussi divers que la photochimie, la biologie ou les sciences des matériaux.
Référence :
Local NH-π interactions involving aromatic residues of proteins: influence of backbone conformation and ππ* excitation on the π H-bond strength, as revealed from studies of isolated model peptides, W. Y. Sohn, V. Brenner, E. Gloaguen, M. Mons, M. Phys. Chem. Chem. Phys. 2016, 18, 29969

Trajectoire de dynamique non-adiabatique dans un peptide contenant une phénylalanine
• ![]() Interaction laser-matière › Physico-Chimie et Chimie-Physique
Interaction laser-matière › Physico-Chimie et Chimie-Physique
• Interactions, Dynamics and Lasers Laboratory (LIDYL) - CEA-CNRS and Paris Saclay University • Laboratoire Interactions, Dynamiques et Lasers (LIDYL) - CEA-CNRS et Université Paris Saclay
• Group "BioMolecular Structures" • LUMO-SBM : Equipe Structures BioMoléculaires
