• ![]() Interaction laser-matière › Physico-Chimie et Chimie-Physique
Interaction laser-matière › Physico-Chimie et Chimie-Physique
• Laboratoire Interactions, Dynamiques et Lasers (LIDYL) - CEA-CNRS et Université Paris Saclay
• LUMO-DyR - Reaction Dynamics Team • LUMO - Equipe Dynamique Réactionnelle (DyR)

Photodepletion measurements of the Zr center dot center dot center dot F-CH3 van der Waals complex
S. Soorkia, J.-M. Mestdagh, M.-A. Gaveau and B. Soep
Chem. Phys. Lett., 491, 140, 2010 [doi]
We report here on the observation of the Zr⋯F–CH3 van der Waals complexes in a laser ablation – supersonic expansion source. The complexes are studied using a combination of laser spectroscopy and time-of-flight mass spectrometric detection. We show that the (1:1) Zr⋯F–CH3 complex is stable in the ground electronic state. By a ground state photodepletion method, we have characterized one of the transitions in the 600–700 nm region as correlating asymptotically with the atomic transition from the ground state of free Zr atom. The corresponding excited state of the Zr⋯F–CH3 complex is reactive with activation of the C–F bond.

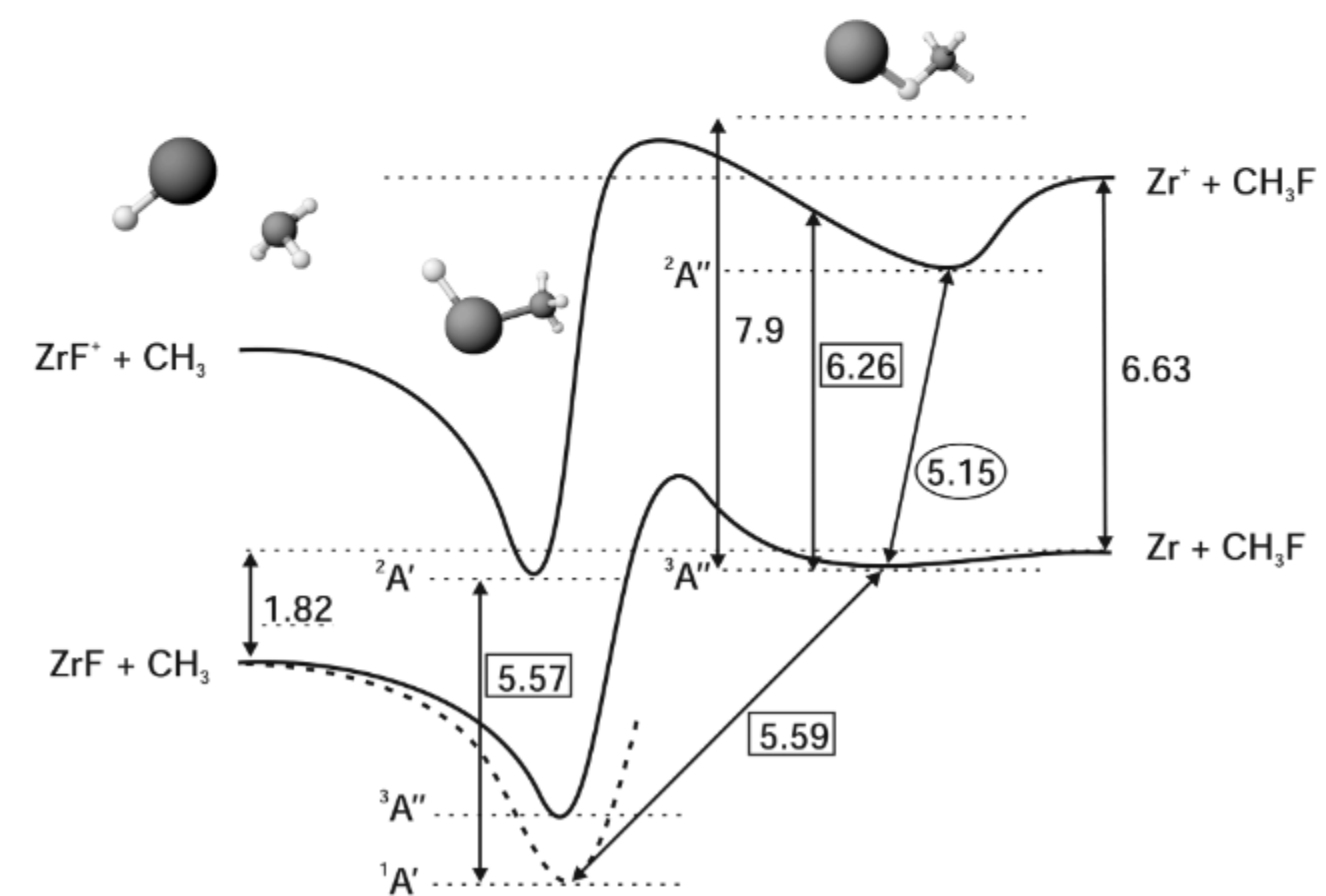
Reactions of Laser-Ablated Zirconium Atoms within a Supersonic Expansion: Insertion versus Radical Mechanism
S. Soorkia, C. Pothier, M. Mestdagh, B. Soep and J. Lievin
J. Phys. Chem. A, 114, 5655, 2010 [doi]
In a laser ablation type microreactor followed by supersonic expansion, zirconium atoms have been reacted with methyl fluoride, CH3F (MeF), and mixtures of MeF and dimethylether, CH3−O−CH3 (DME) seeded in He. With both mixtures, only a number of simple fluorinated products are formed, and they have been identified by one-photon ionization. All products can be linked to radical reactions either with F atoms, CH3, or ZrF1, 2, 3 radicals. No insertion products of the Grignard reagent type, F −Zr−CH3 could be identified with or in the absence of DME. On the other hand, evidence has been found for the presence of organometallic compounds of the type ZrC2Hn=2, 4, 6, which could result from radical attack. Thus, even in conditions where intense solvation is at work, induced by clustering with polar DME molecules, which can act as stabilizing agents, a direct insertion mechanism into the C−F bond involving barrier suppression is not at work in our conditions. The reactivity due to radicals is very effective in this type of reactor, and the products that are efficiently formed can be quickly stabilized in the expansion. The radical attack supersedes, in the case of zirconium solvated by DME, the metastable mechanism with Zr(4d)3(5s)1, that is certainly energetically impossible in the absence of strong reaction barrier suppression by a solvent. High level ab initio calculations performed at the CASPT2 level of theory are used for characterizing the electronic and geometric structure of the inserted products. They also reveal striking features of the reaction mechanism that support the absence of observation of inserted products within solvated clusters of zirconium.
Determination of the Ground Electronic State in Transition Metal Halides: ZrF
S. Soorkia, N. Shafizadeh, J. Lievin, M.-A. Gaveau, C. Pothier, J.-M. Mestdagh, B. Soep and R. W. Field
J. Phys. Chem. A, 115, 9620, 2011 [doi]
The spectroscopy of the ZrF radical, produced by a laser ablation–molecular beam experimental setup, has been investigated for the first time using a two-color two-photon (1 + 1′) REMPI scheme and time-of-flight (TOF) mass spectrometry detection. The region of intense bands 400–470 nm has been studied, based upon the first spectroscopic observations of the isovalent ZrCl radical by Carroll and Daly.(1) The overall spectrum observed is complex. However, simultaneous and individual ion detection of the five naturally occurring isotopologues of ZrF has provided a crucial means of identifying band origins and characterization via the isotopic shift, δiso, of the numerous vibronic transitions recorded. Hence, five (0–0) transitions, of which only two were free of overlap with other transitions, have been identified. The most intense (0–0) transition at 23113 cm–1 presented an unambiguously characteristic RQP rotational structure. From rotational contour simulations of the observed spectra, the nature of the ground electronic state is found to be unambiguously of 2Δ symmetry, leading to the assignment of this band as (0–0) 2Δ ← X2Δ at 23113 cm–1. A set of transitions (1–0) 2Δ ← X2Δ at 22105 cm–1 and (2–0) 2Φ ← X2Δ at 22944 cm–1 involving the X2Δ state has also been identified and analyzed. Furthermore, a second series of transitions with lesser intensity has also been related to the long-lived metastable 4Σ– state: (3–0) 4Π–1/2 ← 4Σ– at 21801 cm–1, (2–0) 4Π–1/2 ← 4Σ– at 21285 cm–1 and (2–0) 4Σ– ← 4Σ– at 23568 cm–1. These spectroscopic assignments are supported by MRCI ab initio calculations, performed using the MOLPRO quantum chemistry package, and show that the low-lying excited states of the ZrF radical are the 4Σ– and 4Φ states lying at 2383 and 4179 cm–1 respectively above the ground X 2Δ state. The difference in the nature of ground state and ordering of the first electronic states with TiF (X 4Φ)(2-4) and ZrCl,(5) respectively, is examined in terms of the ligand field theory (LFT)(7) applied to diatomic molecules. These results give a precise description of the electronic structure of the low lying electronic states of the ZrF transition metal radical.

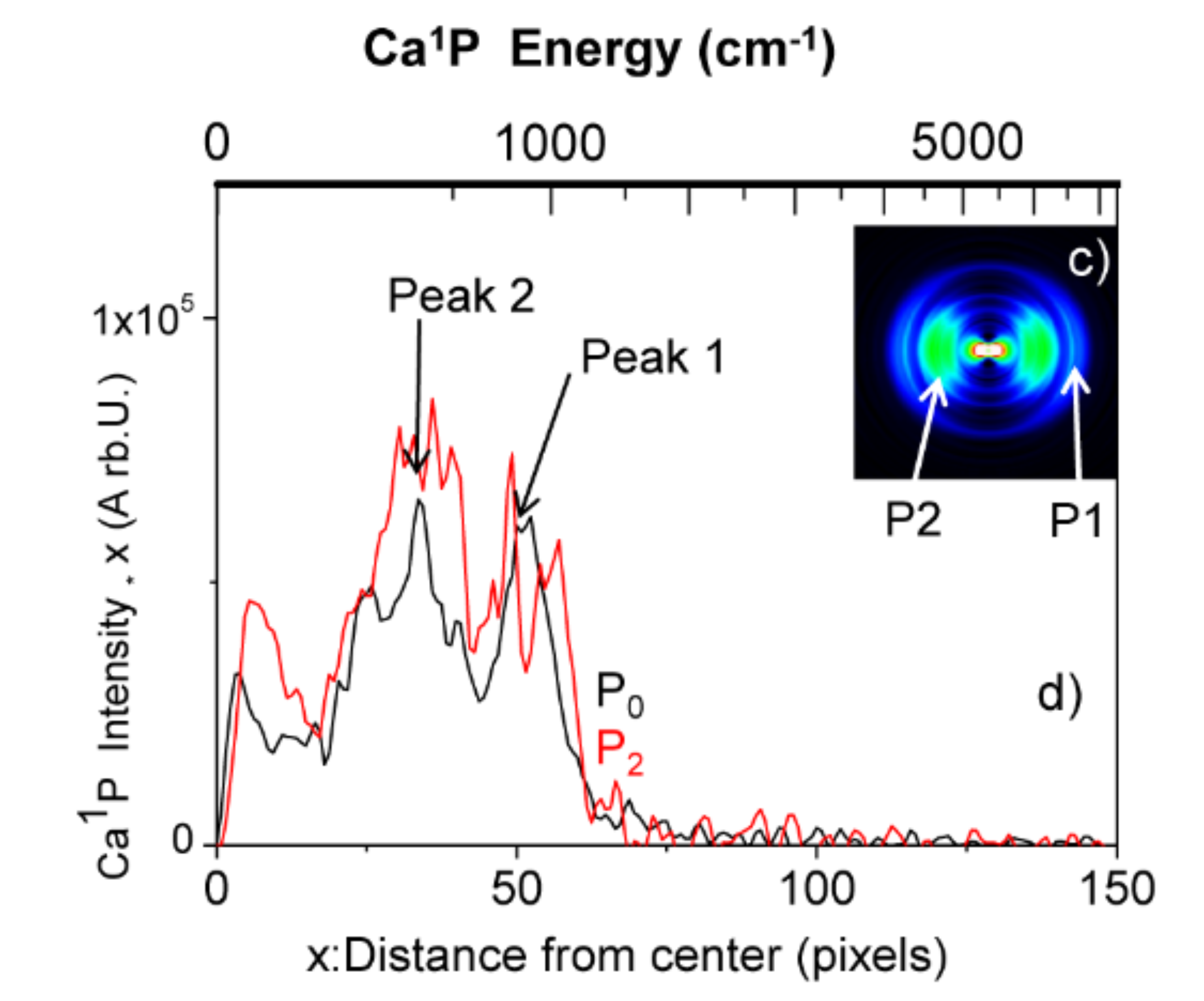
Reactive and Inelastic Channels in the Ca*···FCH3 Transition State: A Simple Branching Mechanism
M. Briant, E. Gloaguen, A. Beswick, J. M. Mestdagh, S. Stolte, L. Poisson, C. Pothier and B. Soep
J. Phys. Chem. A, 2015 [doi]
To study the excited state dynamics between a calcium atom and the CH3F molecule, a Ca center dot center dot center dot CH3F 1:1 complex has been prepared by a supersonic expansion with laser ablation of calcium metal in the gas phase. Tunable laser excitation of these complexes in molecular states correlating to Ca P-1(1)(4s4p) + CH3F allows observing two competitive channels: the direct dissociation and the reactive channel into CaF* + CH3. The translational recoil, as well as the alignment of the fragments Ca* and CaF* have been analyzed by velocity map imaging and time-of-flight mass spectrometry. This revealed that both the dissociation and reaction processes are quasi direct and are of comparable intensity. We provide a simple interpretation for this process: the electronically excited potential surface of the Ca*center dot center dot center dot FCH3 complex initiates a fast predissociation from a suspended well to two repulsive surfaces that lead either to Ca P-1(1)(4s4p) (Omega = 1) + CH3F or to CaF((2)Delta) + CH3.
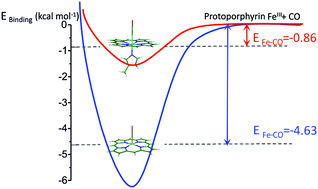
The dramatic effect of N-methylimidazole on trans axial ligand binding to ferric heme: experiment and theory
M. Aarabi, R. Omidyan, S. Soorkia, G. Grégoire, M. Broquier, M.-E. Crestoni, A. de la Lande, B. Soep and N. Shafizadeh
Phys. Chem. Chem. Phys., 21, 1750, 2019 [doi]
The binding energy of CO, O2 and NO to isolated ferric heme, [FeIIIP]+, was studied in the presence and absence of a σ donor (N-methylimidazole and histidine) as the trans axial ligand. This study combines the experimental determination of binding enthalpies by equilibrium measurements in a low temperature ion trap using the van’t Hoff equation and high level DFT calculations. It was found that the presence of N-methylimidazole as the axial ligand on the [FeIIIP]+ porphyrin dramatically weakens the [FeIIIP–ligand]+ bond with an up to sevenfold decrease in binding energy owing to the σ donation by N-methylimidazole to the FeIII(3d) orbitals. This trans σ donor effect is characteristic of ligation to iron in hemes in both ferrous and ferric redox forms; however, to date, this has not been observed for ferric heme.