








[Gd(HP-DO3A)] contrast agent in aqueous solution
Medical imaging is an invaluable tool for the diagnosis of cancer especially when non-invasive techniques based on nuclear magnetic resonance are used. Their high spatial resolution stems from metal-based contrast agents which are mostly Gd(III) chelates. In spite of the prevalence of these paramagnetic contrast agents in oncology, improvements are awaited regarding their efficiency, their selectivity, and even their toxicity. Both experimental and theoretical chemists can contribute to understand the mechanism of their enhancing power and to suggest innovative contrast agents that will replace the existing ones.
Presently, atomic scale simulations of metal complexes in solution such as Gd-based contrast agents can essentially be divided in high-level quantum chemistry calculations or molecular dynamics simulations. More recent theoretical approaches could be considered as the “best of both worlds”. These are ab initio simulations launched by Car and Parrinello in 1985 but applied much later for chemistry applications. The study of Gd chelates in aqueous solution (namely ProHance®), clinically approved as MRI contrast agents, using ab initio molecular dynamics (AIMD) method was first reported by our team in 2007 [1].
Although both AIMD simulations represented a real step forward in modeling MRI contrast agents, they both suffered from statistics errors due to the too short trajectories. The length of such Car-Parrinello simulations is indeed typically limited to a few dozen picoseconds, which can be enough to compute average properties but too short to observe some of the events which are relevant for MRI. For example, the water exchange rate between the first and the second coordination spheres in commercial Gd-based MRI contrast is in the range of few hundreds nanoseconds. Observing such event is therefore completely out of reach during standard molecular dynamics simulations (even classical ones).
The use of accelerated techniques can however boost the sampling of rare events. The first accelerated ab initio molecular dynamics simulations on MRI contrast agents was reported by our team in 2011. Here the metadynamics method was used for the study of the water exchange reaction in two isomers of the ProHance® contrast agent [2]. The method was coupled to ab initio molecular dynamics, providing accurate free-energy profiles along the reaction coordinate from which free-energy barriers were extracted.
Beside these theoretical studies of chemical mechanisms, our ab initio simulations have been used to analyse the fluctuations of magnetic properties, such as as hyperfine interactions (between electronic and nuclear spins) [3] and zero field splitting (for electronic spin relaxation) [4,5] along a trajectory.
Also, these ab initio simulations, combining Car-Parrinello molecular dynamics and metadynamics, have been applied to contrast agents based on a completely different physical mechanism, namely PARACEST agents (for Paramagnetic Chemical Exchange Saturation Transfer) [6].
[1] R. Pollet and D. Marx, J. Chem. Phys. 126 181102 (2007) (download)
Copyright 2007 American Institute of Physics. This article may be downloaded for personal use only. Any other use requires prior permission of the author and the American Institute of Physics.
[2] R. Pollet, N. Nair, D. Marx, Inorg. Chem. 50 4791 (2011)
[3] A. Lasoroski, R. Vuilleumier, R. Pollet, J. Chem. Phys. 139 104115 (2013)
[4] A. Lasoroski, R. Vuilleumier, R. Pollet, J. Chem Phys. 140 014201 (2014)
[5] S. Khan, R. Pollet, R. Vuilleumier, J. Kowaleswski, M. Odelius, J. Chem. Phys. 147 244306 (2017)
[6] R. Pollet, C. S. Bonnet, P. Retailleau, P. Durand, E. Toth, Inorg. Chem. 56 4317 (2017)
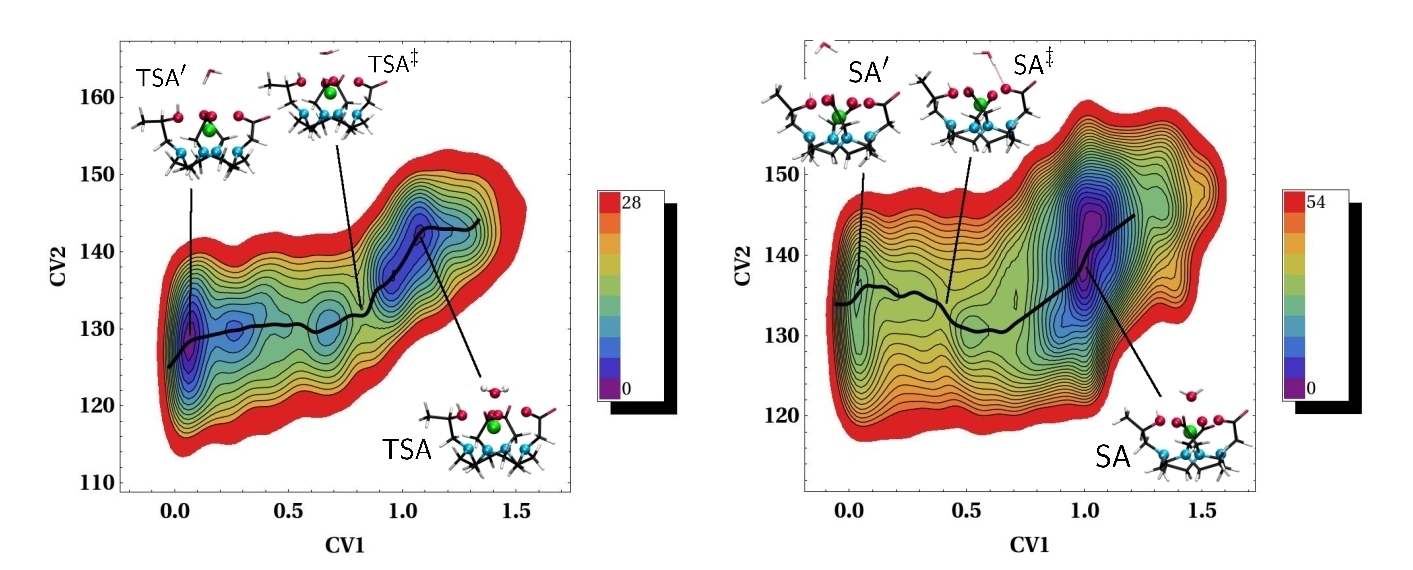
Free energy surfaces for the water exchange reaction for two isomers of the Prohance contrast agent
• ![]() Materials for new energy technologies › Quantum chemistry and molecular simulations
Materials for new energy technologies › Quantum chemistry and molecular simulations
• Service Interdisciplinaire sur les Systèmes Moléculaires et les Matériaux
