Contact : Michel Mons
L’équipe LUMO-SBM, regroupée avec l’équipe LUMO-DyR, pour former le groupe de recherche LUMO, est historiquement liée à l’application des lasers à la physico-chimie moléculaire,et tournée vers l’interaction des systèmes moléculaires complexes avec la lumière. Elle étudie notamment (mais pas exclusivement) des molécules neutres, modèles de biomolécules, isolées en phase gazeuse. L’objectif scientifique général est de documenter, par diverses spectroscopies laser et par modélisation théorique, les interactions rencontrées dans ces systèmes, aussi bien dans leur état fondamental que dans leurs états électroniques excités. L’un des enjeux est alors de résoudre la complexité conformationnelle, tautomérique, etc… de ces objets afin de pouvoir documenter le rôle de la structure sur leurs propriétés dynamiques, notamment leur dynamique électronique, au travers d’expériences de type pompe-sonde. Ce programme est réalisé grâce à une forte synergie expérience-théorie assurée par la présence simultanée de ces deux compétences au sein de l’équipe, constituée de chimistes et de physiciens.
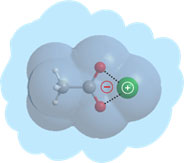
L’essentiel de l’activité se partage entre trois thèmes principaux, liés à la structure et la dynamique électronique de systèmes flexibles et de biomolécules, et une action de valorisation.
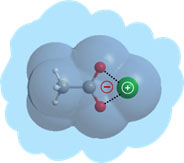
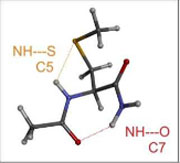
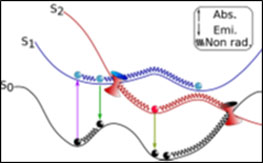

L’équipe « Structures BioMoléculaires » est implantée au Bât. 522 du CEA Saclay. Elle maintient son propre parc de lasers nanoseconde et utilise les lasers femtoseconde/picoseconde des serveurs LUCA/ATTOLAB à Saclay et CLUPS à l’Université Paris-Sud ainsi que le Synchrotron SOLEIL. Enfin, elle maintient son propre parc de stations de travail (~ 4 noeuds/50 procs) et utilisent les ressources nationales du « Calcul Intensif » (TGIR GENCI & CCRT-CEA).