








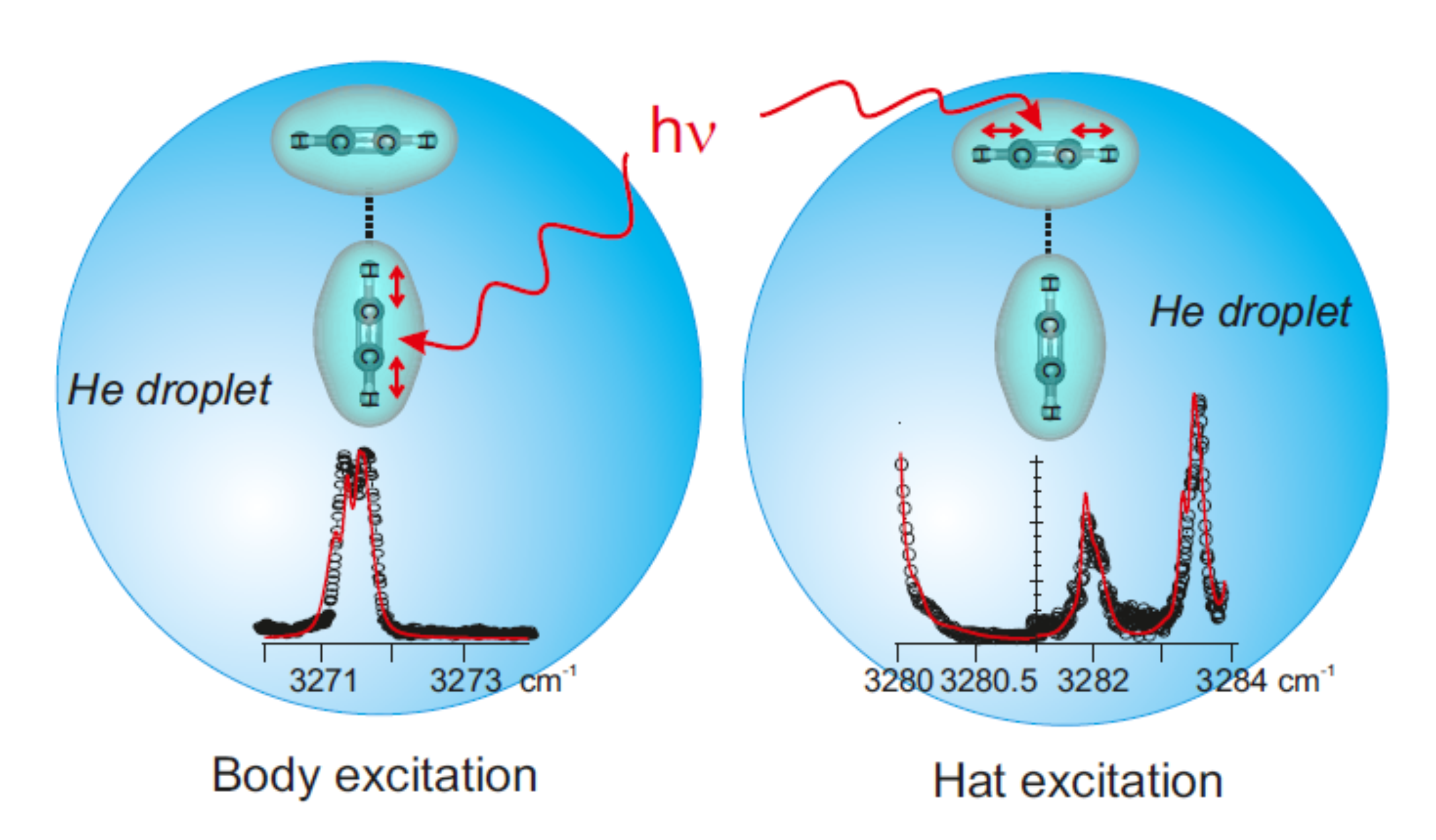
Dynamics of acetylene dimers hosted in helium droplets
M. Briant, E. Mengesha, M. A. Gaveau, B. Soep, J. M. Mestdagh and L. Poisson
Phys. Chem. Chem. Phys., 20, 2597, 2018 [doi]
The CH antisymmetric stretch of the C2H2 moieties in acetylene dimers was explored over the range 3270-3290 cm-1 using the helium nanodroplet isolation (HENDI) technique. This work is part of a general investigation which addresses the dynamical consequences of coupling the deformation motions of weakly bound complexes with a finite size quantum liquid (the helium droplet). The acetylene dimer is attractive from this point of view because one of its deformation coordinates promotes a tunneling isomerization process. A numerical simulation of the observed spectrum allows deriving a set of effective spectroscopic constants which help understanding the dynamical role played by the droplet on the rotation and deformation of the dimer.
A HElium NanoDroplet Isolation (HENDI) investigation of the weak hydrogen bonding in the propyne dimer (CH3CCH)2
A. Gutiérrez-Quintanilla, M. Briant, E. Mengesha, M. A. Gaveau, J. M. Mestdagh, B. Soep, C. Crépin and L. Poisson
Phys. Chem. Chem. Phys., 20, 28658, 2018 [doi]
A HElium Nanodroplet Isolation (HENDI) experiment was performed to explore the absorption spectra of the propyne monomer (CH3CCH), dimer and (CH3CCH)≥3 multimers in the vicinity of the CH stretch region ν1 of the monomer. Ab initio calculations were performed at the MP2 level to document the potential energy surface of the dimer. This provided the necessary parameters to simulate the absorption spectrum of the dimer and thus facilitate the interpretation of the experiment. The central result was to observe three isomers of the dimer, hence reflecting the complexity of the weak CH⋯π H-bonding when several H-donors are at play.
Propyne-water complexes hosted in helium droplets
A. Gutiérrez-Quintanilla, M. Briant, E. Mengesha, M.-A. Gaveau, J.-M. Mestdagh, B. Soep and L. Poisson
Low Temp. Phys., 45, 634, 2019 [doi]
A HElium Nanodroplet Isolation (HENDI) experiment was performed to explore the absorption spectrum of the propyne-water complex (CH3CCH-H2O). Two spectral regions were investigated, near the CH stretch v1 of the propyne moiety and near the asymmetric stretch v3 of the water moiety. Ab-initio calculations were performed at the MP2/aug-cc-pVTZ level to estimate the spectroscopic constants of the free complex. This provided the necessary parameters to simulate the absorption spectrum of the complex and thus facilitate the interpretation of the experiment. The observed spectrum is consistent with a structure of the complex where two H-bonds between water and propyne form a five member ring. The later was predicted by Lopes et al. [J. Mol. Struct. 834, 258 (2007)].
• ![]() Laser-matter interaction › Physico-chemistry and Chemical-physics
Laser-matter interaction › Physico-chemistry and Chemical-physics
• LUMO-DyR - Reaction Dynamics Team • LUMO - Equipe Dynamique Réactionnelle (DyR)
• Isolation d'une réaction chimique dans un agrégat de grande taille. Technique CICR.
