








Ultrafast electronic relaxation of excited state vitamin B12 in the gas phase
N. Shafizadeh, L. Poisson and B. Soep
Chem. Phys., 350, 2, 2008 [doi]
The time evolution of electronically excited vitamin B12 (cyanocobalamin) has been observed for the first time in the gas phase. It reveals an ultrafast decay to a state corresponding to metal excitation. This decay is interpreted as resulting from a ring to metal electron transfer. This opens the observation of the excited state of other complex biomimetic systems in the gas phase, the key to the characterisation of their complex evolution through excited electronic states.
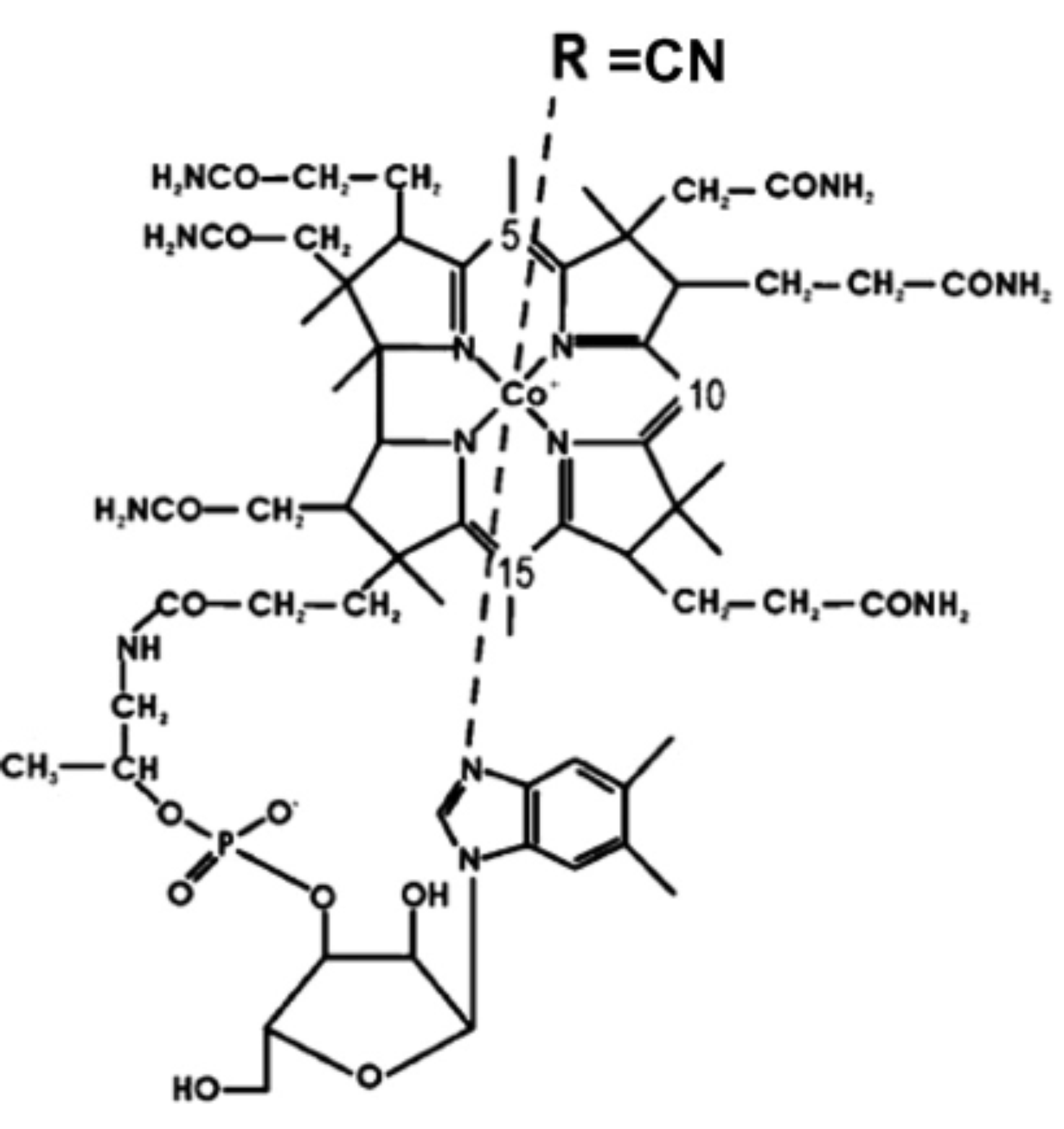
Charge transfer in metal-atom-containing molecules in the gas phase
N. Shafizadeh, B. Soep, J. M. Mestdagh and W. H. Breckenridge
Int. Rev. Phys. Chem., 28, 359, 2009 [doi]
The presence of charge transfer (CT) is ubiquitous in metal-atom-containing molecules in the gas phase. The states of interest range from ground states to the lower excited states, given the generally low ionisation potentials of metal atoms. This review is written from an experimentalist's perspective, to describe the specifics of CT states in stable molecules containing a metal atom or of transient CT states in dynamical processes. The large body of experimental evidence allows the description of such states in an empirical or semi-empirical manner, including partial point charges and polarised electron clouds on the ions. These simple models are compared with more quantitative models, allowing the development of sophisticated descriptions. The simple perspective deriving from experimental evidence is justified, when it is possible via accurate quantum calculations and allows building a rather complete framework for the ionic bond or CT-induced dynamics.
First observation in the gas phase of the ultrafast electronic relaxation pathways of the S-2 states of heme and hemin
M.-H. Ha-Thi, N. Shafizadeh, L. Poisson and B. Soep
Phys. Chem. Chem. Phys., 12, 14985, 2010 [doi]
The time evolution of electronically excited heme (iron II protoporphyrin IX, [Fe-II PP]) and its associated salt hemin (iron III protoporphyrin IX chloride, [Fe-III PP-Cl]), has been investigated for the first time in the gas phase by femtosecond pump-probe spectroscopy. The porphyrins were excited at 400 nm in the S-2 state (Soret band) and their relaxation dynamics was probed by multiphoton ionization at 800 nm. This time evolution was compared with that of the excited state of zinc protoporphyrin IX [Zn PP] whose S2 excited state likely decays to the long lived S-1 state through a conical intersection, in less than 100 fs. Instead, for [Fe-II PP] and [Fe-III PP-Cl], the key relaxation step from S-2 is interpreted as an ultrafast charge transfer from the porphyrin excited orbital pi* to a vacant d orbital on the iron atom (ligand to metal charge transfer, LMCT). This intermediate LMCT state then relaxes to the ground state within 250 fs. Through this work a new, serendipitous, preparation step was found for Fe II porphyrins, in the gas phase.
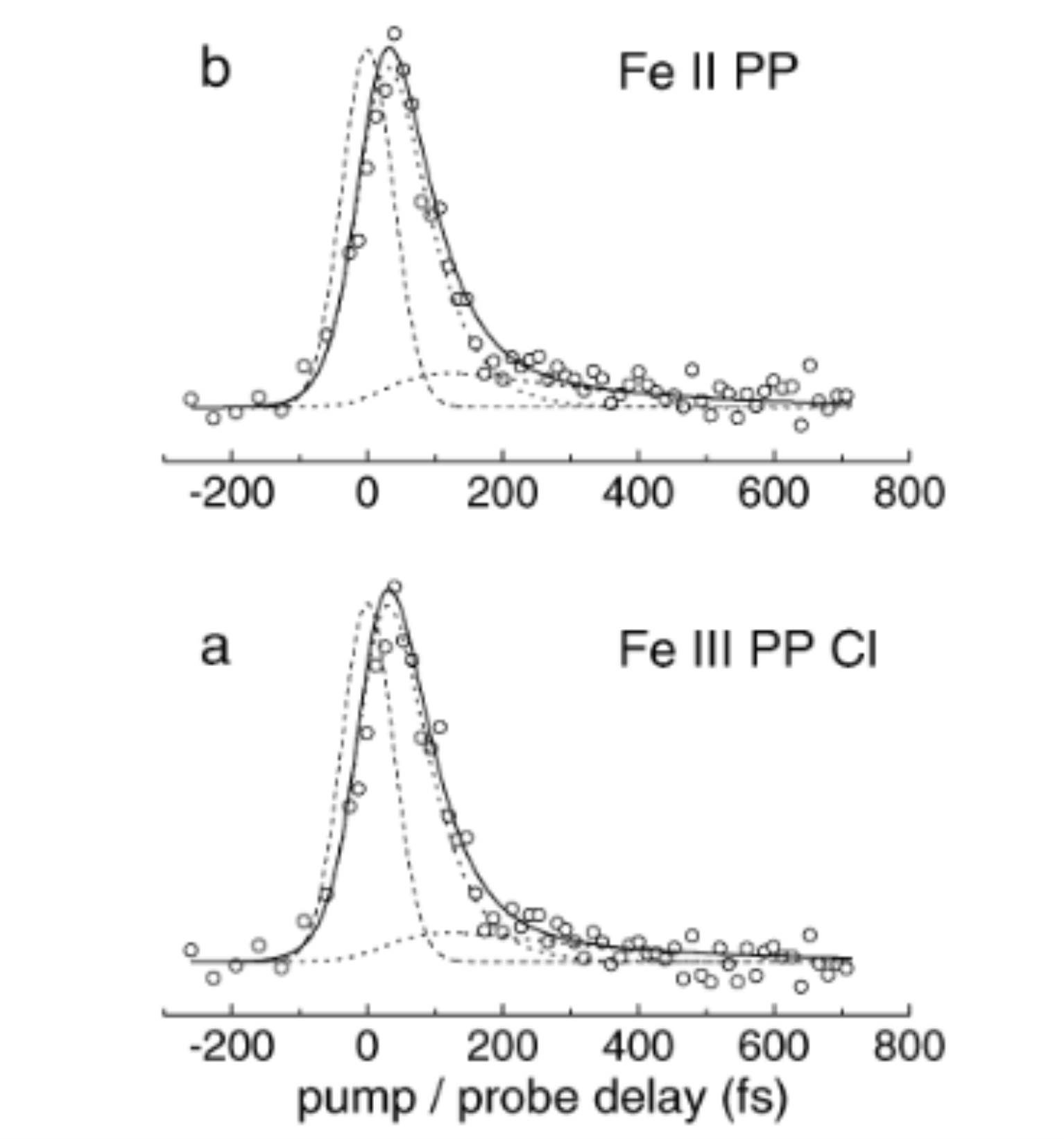
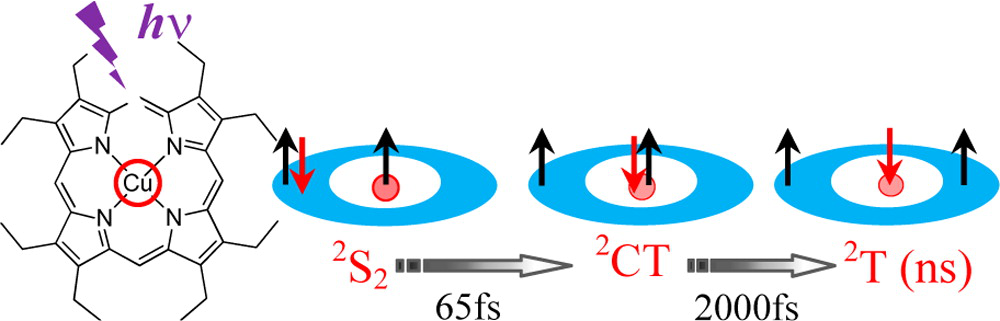
An Efficient Indirect Mechanism for the Ultrafast Intersystem Crossing in Copper Porphyrins
M.-H. Ha-Thi, N. Shafizadeh, L. Poisson and B. Soep
J. Phys. Chem. A, 117, 8111, 2013 [doi]
The ultrafast dynamics of copper tetraphenylporphyrin (CuTPP), copper octaethylporphyrin (CuOEP), and of the free base tetraphenylporphyrin (H2TPP), excited in the S2 state have been investigated in the gas phase by femtosecond pump/probe experiments. The porphyrins were excited in the Soret band at 400 nm. Strikingly, the S2–S1 internal conversion in H2TPP is very rapid (110 fs), as compared to that of ZnTPP (600 fs), previously observed. In turn, CuTPP and CuOEP, excited in S2, follow an efficient and different relaxation pathway from that of other open-shell metalloporphyrins. These two molecules exhibit a sequential four-step decay ending on a slow evolution in the nanosecond range 2S2 → 2CT → 2T → 2Ground State. This latter evolution is linked to the formation of the 2T, tripdoublet state in CuTPP, observed in the condensed phase. It is shown that an intermediate charge transfer state plays a crucial role in linking the porphyrin centered 1ππ* and 3ππ* configurations. A simple model is presented that allows a rapid evolution between these two configurations, via coupling of the porphyrin π system with the free d electron on the copper. The mechanism obviates the need for the spin orbit coupling within the porphyrin. The result is that these copper porphyrins can exhibit an ultrafast apparent intersystem crossing, unprecedented for organic molecules.
Observation in the gas phase of the ligation of 1-Methylimidazole to hemoprotein mimics
N. Shafizadeh, M. H. Ha-Thi, L. Poisson, B. Soep and P. Maillard
J. Chem. Phys., 141, 174310, 2014 [doi]
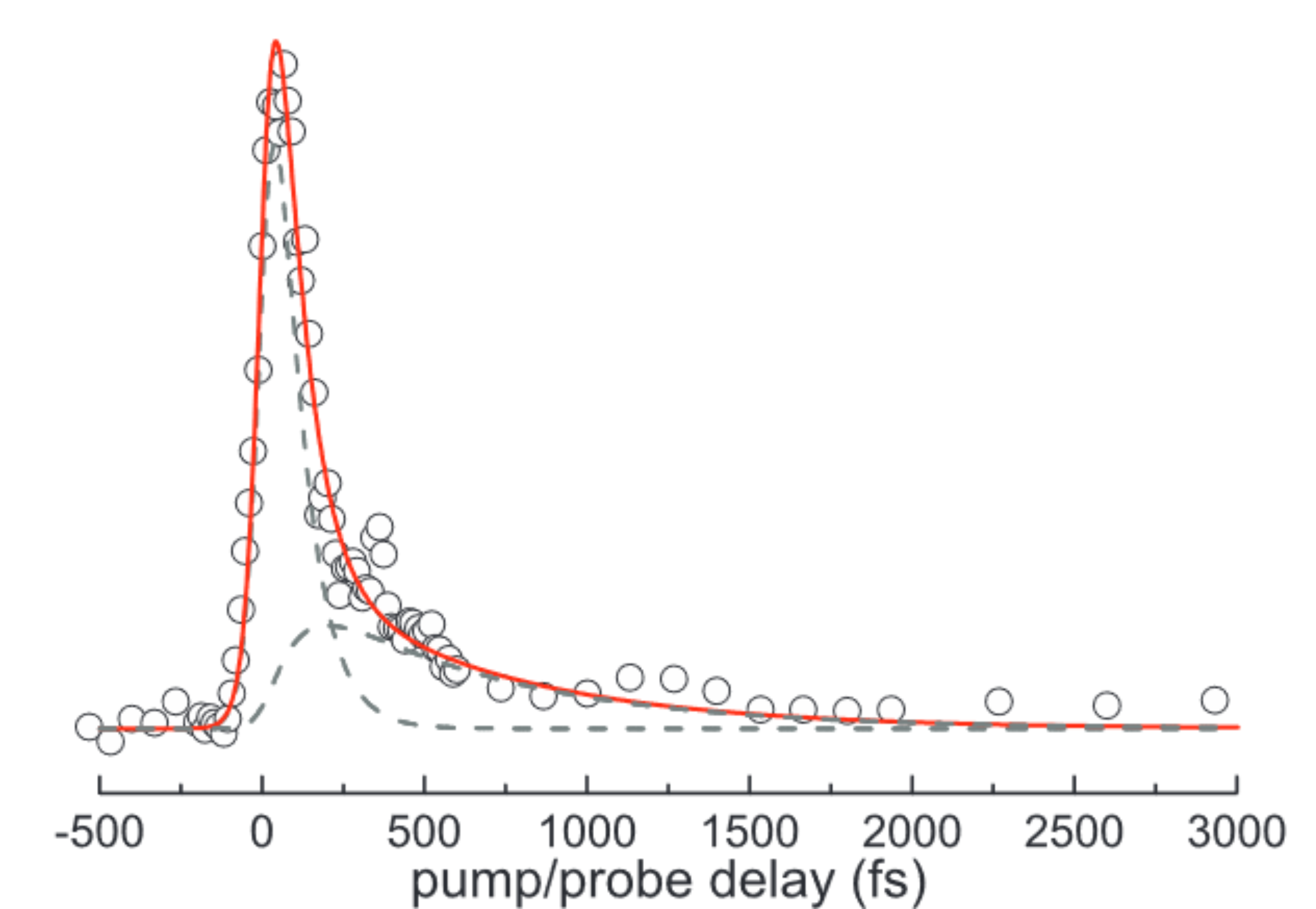
Hemoprotein mimics, cobalt picket fence porphyrins have been prepared in the gas phase as neutral molecules for the first time. Their ligation properties have been studied with 1-methylimidazole and compared with those of other cobalt porphyrins, tetraphenyl porphyrin, and cobalt protoporphyrin IX chloride, in view of studying the sterical properties of the ligation. It is shown that the cobalt picket fence porphyrin can only accept one 1-methylimidazole ligand in contrast to less sterically crowded porphyrins like cobalt tetraphenylporphyrin that present two accessible ligation sites. The femtosecond dynamics of these ligated systems have been studied after excitation at 400 nm, in comparison with the unligated ones. The observed transients are formed in much shorter times, 30 fs for the ligated species, as compared to free species (100 fs), supporting the porphyrin to metal charge transfer nature of these transients. The similar decays of the ligated transients <1 ps reveal the absence of photodissociation of the cobalt-1-methylimidazole bond at this step of evolution. (C) 2014 AIP Publishing LLC.
Structure of cobalt protoporphyrin chloride and its dimer, observation and DFT modeling
A. de la Lande, M.-H. Ha-Thi, S. Chen, B. Soep and N. Shafizadeh
Phys. Chem. Chem. Phys., 18, 16700, 2016 [doi]
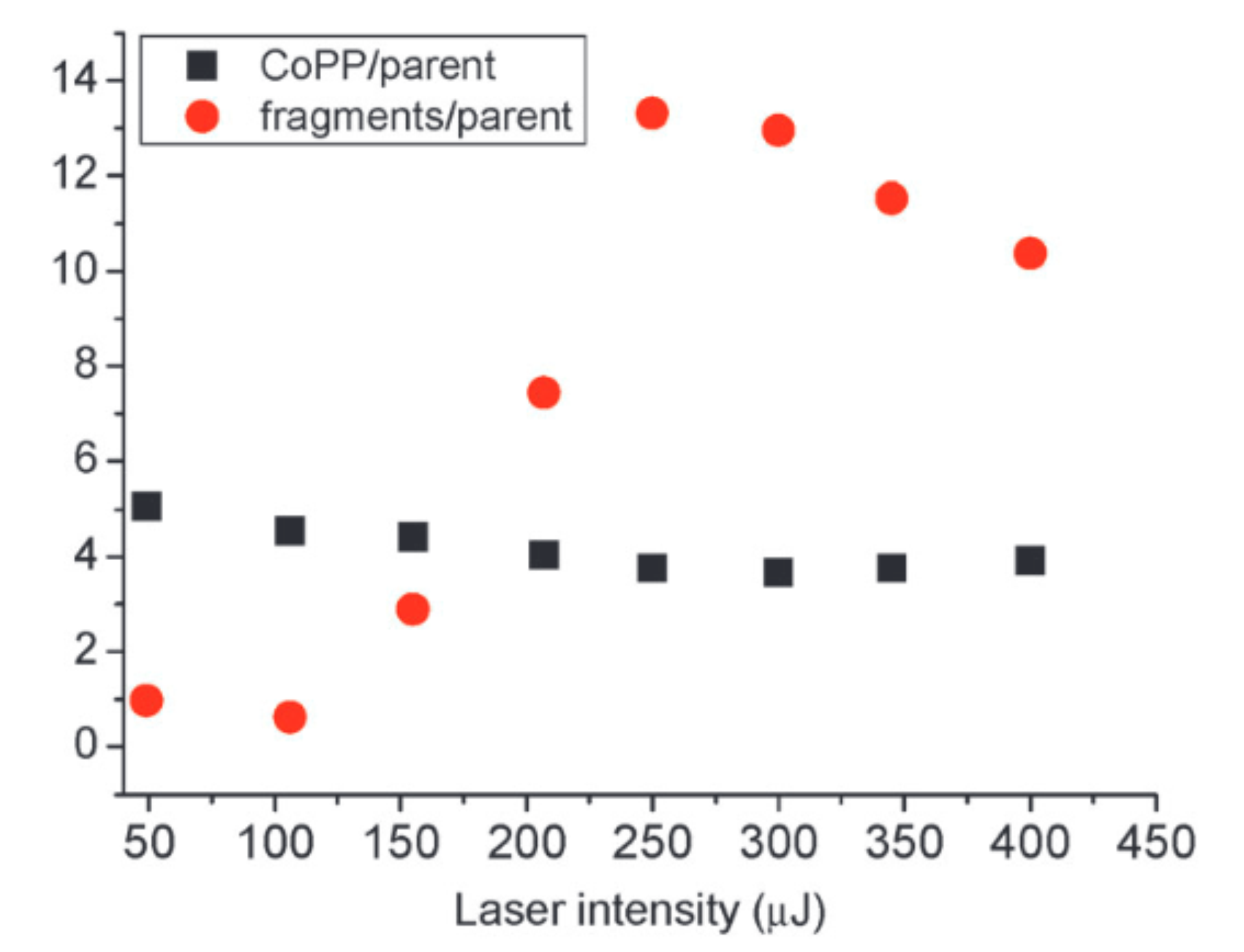
In this article we present a joint study by time-of-flight mass spectroscopy and density functional theory of cobalt protoporphyrin dimer complexes. The main novelty of the experimental part is to reveal the formation of porphyrin dimers that eventually include a chlorine atom. Density functional theory calculations have been performed to shed light on the structural and electronic properties of monomers and dimers that may be formed experimentally. Various geometries of the monomers are analyzed in the two lowest spin states. The electronic structures are examined by means of population analysis relying on the iterative Hirshfeld scheme and the topological analyses of the electron localization function. It is shown that the cobalt ligand bond is purely ionic in the triplet states but shows a noticeable covalent character in the singlet state. Ionization potential of Co-protoporphyrin and binding energies of the chlorine ligand are also reported. Concerning the dimers, several association patterns are investigated for the chlorinated and non-chlorinated complexes. It is found that the structures of the most stable complexes involve four hydrogen bonds between the carboxylic acid moieties of the protoporphyrins. However other association modes are likely to be possible in the experiments.
• ![]() Laser-matter interaction › Physico-chemistry and Chemical-physics
Laser-matter interaction › Physico-chemistry and Chemical-physics
• Laboratoire Interactions, Dynamiques et Lasers (LIDYL) - CEA-CNRS et Université Paris Saclay
• LUMO-DyR - Reaction Dynamics Team • LUMO - Equipe Dynamique Réactionnelle (DyR)
