








Experimental Evidence for Ultrafast Electronic Relaxation in Molecules, Mediated by Diffuse States
E. Gloaguen, J. M. Mestdagh, L. Poisson, F. Lepetit, J. P. Visticot, B. Soep, M. Coroiu, A. T. J. B. Eppink and D. H. Parker
J. Am. Chem. Soc., 127, 16529, 2005 [doi]
The existence of a mediating state for the ultrafast electronic relaxation in ethylenic-like molecules has been shown by femtosecond electron imaging techniques. This state is of Rydberg character, and its high efficiency is due to its ability to link the electron distributions of the initial and final electronic states.


Time-resolved photoion and photoelectron imaging of NO2
N. T. Form, B. J. Whitaker, L. Poisson and B. Soep
Phys. Chem. Chem. Phys., 8, 2925, 2006 [doi]
Time-resolved photoion and photoclectron velocity mapped images from NO2 excited close to its first dissociation limit [to NO(X-2 Pi) + O(P-3(2))] have been recorded in a two colour pump-probe experiment, using the frequency-doubled and frequency-tripled output of a regeneratively amplified titanium-sapphire laser. At least three processes are responsible for the observed transient signals; a negative pump-probe signal (corresponding to a 266 nm pump), a very short-lived transient close to the cross-correlation of the pump and probe pulses but on the 400 nm pump side, and a longer-lived positive pump-probe signal that exhibits a signature of wavepacket motion (oscillations). These transients have two main origins; multiphoton excitation of the Rydberg states of NO2 by both 266 and 400 nm light, and electronic relaxation in the 1(2)B(2) state of NO2, which leads to a quasi-dissociaied NO2 high in the 1(2)A(1) electronic ground state and just below the dissociation threshold. The wavepacket motion that we observe is ascribed to states exhibiting free rotation of the O atom about the NO moiety. These states, which are common for loosely bound systems such as a van der Waals complex but unusual for a chemically-bound molecule, have previously been observed in the frequency domain by optical double resonance spectroscopy but never before in the time domain.
Direct mapping of recoil in the ion-pair dissociation of molecular oxygen by a femtosecond depletion method
A. V. Baklanov, L. M. C. Janssen, D. H. Parker, L. Poisson, B. Soep, J.-M. Mestdagh and O. Gobert
J. Chem. Phys., 129, 214306, 2008 [doi]
Time-resolved dynamics of the photodissociation of molecular oxygen, O(2), via the (3)Sigma(-)(u) ion-pair state have been studied with femtosecond time resolution using a pump-probe scheme in combination with velocity map imaging of the resulting O(+) and O(-) ions. The fourth harmonic of a femtosecond titanium-sapphire (Ti:sapphire) laser (lambda approximate to 205 nm) was found to cause three-photon pumping of O(2) to a level at 18.1 eV. The parallel character of the observed O(+) and O(-) images allowed us to conclude that dissociation takes place on the (3)Sigma(-)(u) ion-pair state. The 815 nm fundamental of the Ti:sapphire laser used as probe was found to cause two-photon electron photodetachment starting from the O(2) ion-pair state, giving rise to (O((3)P)+O(+)((4)S)) products. This was revealed by the observed depletion of the yield of the O(-) anion and the appearance of a new O(+) cation signal with a kinetic energy E(transl)(O(+)) dependent on the time delay between the pump and probe lasers. This time-delay dependence of the dissociation dynamics on the ion-pair state has also been simulated, and the experimental and simulated results coincide very well over the experimental delay-time interval from about 130 fs to 20 ps where two- or one-photon photodetachment takes place, corresponding to a change in the R(O(+),O(-)) interatomic distance from 12 to about 900 A. This is one of the first implementations of a depletion scheme in femtosecond pump-probe experiments which could prove to be quite versatile and applicable to many femtosecond time-scale experiments.
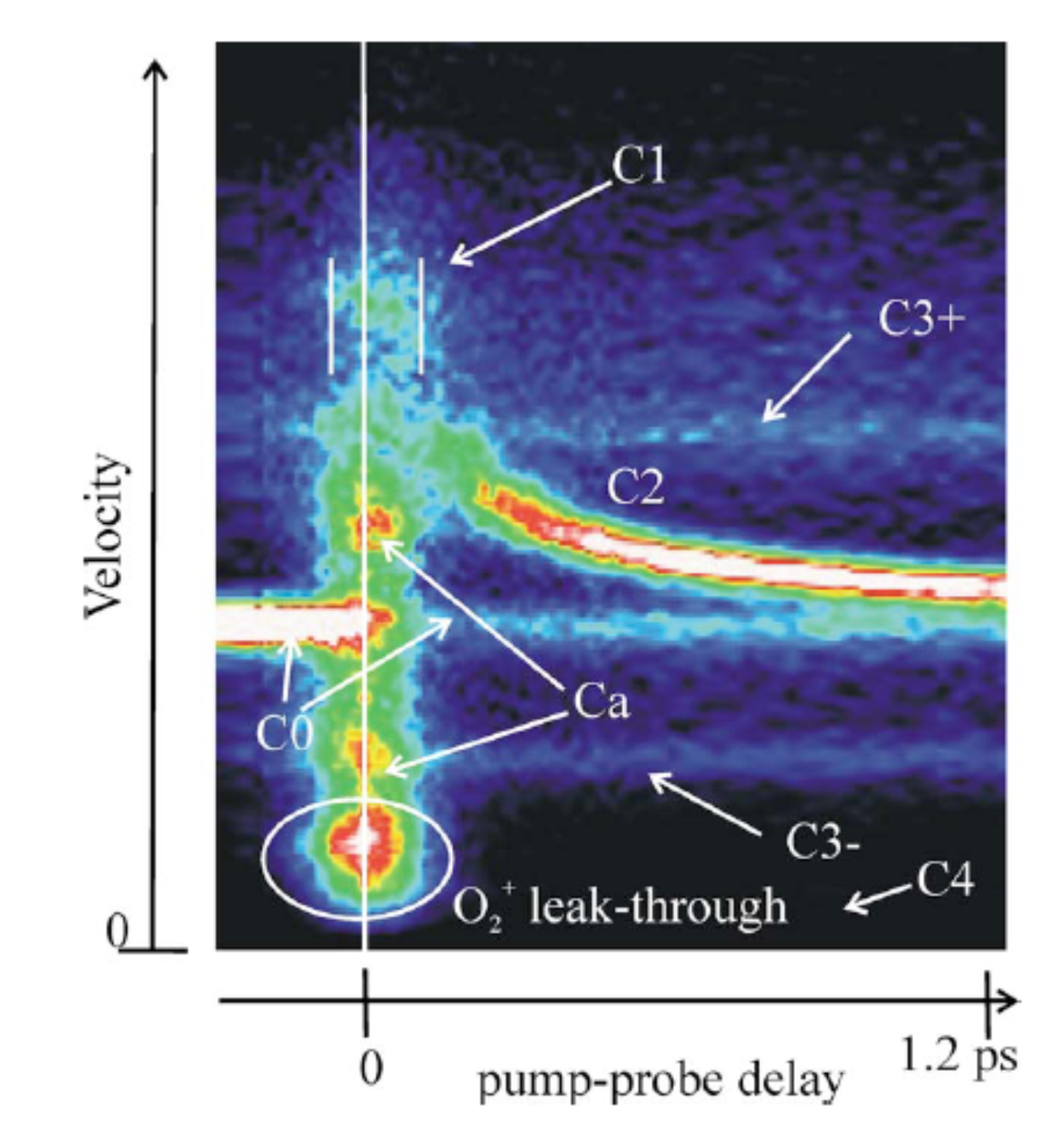

Investigation of ultrafast photoinduced processes for salicylidene aniline in solution and gas phase: toward a general photo-dynamical scheme
M. Sliwa, N. Mouton, C. Ruckebusch, L. Poisson, A. Idrissi, S. Aloise, L. Potier, J. Dubois, O. Poizata and G. Buntinx
Photochemical & Photobiological Sciences, 9, 661, 2010 [doi]
of 2-hydroxybenzylideneaniline (photochromic salicylidene aniline SAOH) and N-(2-methoxybenzylidene) aniline (SAOMe) are studied by steady state and transient optical spectroscopy in solution and gas phase at different excitation wavelengths (266, 355 and 390 nm). Two competitive processes are observed from the enol* excited state: on one hand a rotation to get a twisted-enol, and on the other hand an excited state intramolecular proton transfer (ESIPT) followed by a cis-trans isomerisation to get the trans-keto photochromic product. For the first time both processes are characterized at an ultrashort time scale for salicylidene aniline. Resolution of the spectrokinetic data is achieved by multivariate curve resolution and attribution of the intermediate species recovered is performed in comparison with the results obtained for SAOMe, which can only undergo enol rotational isomerisation. It shows that ESIPT and rotation to the twisted-enol for SAOH occur within 100 fs, as predicted by recent quantum dynamical simulations, with an efficiency ratio dependent on the excitation wavelength. Therefore a general photoinduced mechanism for salicylidene aniline is drawn.
Gas phase dynamics of triplet formation in benzophenone
G. Spighi, M.-A. Gaveau, J.-M. Mestdagh, L. Poisson and B. Soep
Phys. Chem. Chem. Phys., 16, 9610, 2014 [doi]
Benzophenone is a prototype molecule for photochemistry in the triplet state through its high triplet yield and reactivity. We have investigated its dynamics of triplet formation under the isolated gas phase conditions via femtosecond and nanosecond time resolved photoelectron spectroscopy. This represents the complete evolution from the excitation in S2 to the final decay of T1 to the ground state S0. We have found that the triplet formation can be described almost as a direct process in preparing T1, the lowest reacting triplet state, from the S1 state after S2 [rightward arrow] S1 internal conversion. The molecule was also deposited by a pick-up technique on cold argon clusters providing a soft relaxation medium without evaporation of the molecule and the mechanism was identical. This cluster technique is a model for medium influenced electronic relaxation and provides a continuous transition from the isolated gas phase to the relaxation dynamics in solution.

A roaming wavepacket in the dynamics of electronically excited 2-hydroxypyridine
L. Poisson, D. Nandi, B. Soep, M. Hochlaf, M. Boggio-Pasqua and J.-M. Mestdagh
Phys. Chem. Chem. Phys., 16, 581, 2014 [doi]
How much time does it take for a wavepacket to roam on a multidimensional potential energy surface? This combined theoretical and pump-probe femtosecond time experiment on 2-hydroxypyridine proposes an answer. Bypassing the well-established transition state and conical intersection relaxation pathways, this molecular system undergoes relaxation into the S1 excited state: the central ring is destabilized by the electronic excitation, within [similar]100 fs after absorption of the pump photon, then the H-atom bound to oxygen undergoes a roaming behavior when it couples to other degrees of freedom of the molecule. The timescale of the latter process is measured to be [similar]1.3 ps. Further evolution of the wavepacket is either an oscillation onto the S1 potential or a conversion into the triplet state for timescale larger than [similar]110 ps. Our work introduces a new tool for the understanding of time-resolved relaxation dynamics applied to large molecules through the roaming dynamics characterized by its strongly delocalized wavepacket on flat molecular potential energy surfaces.
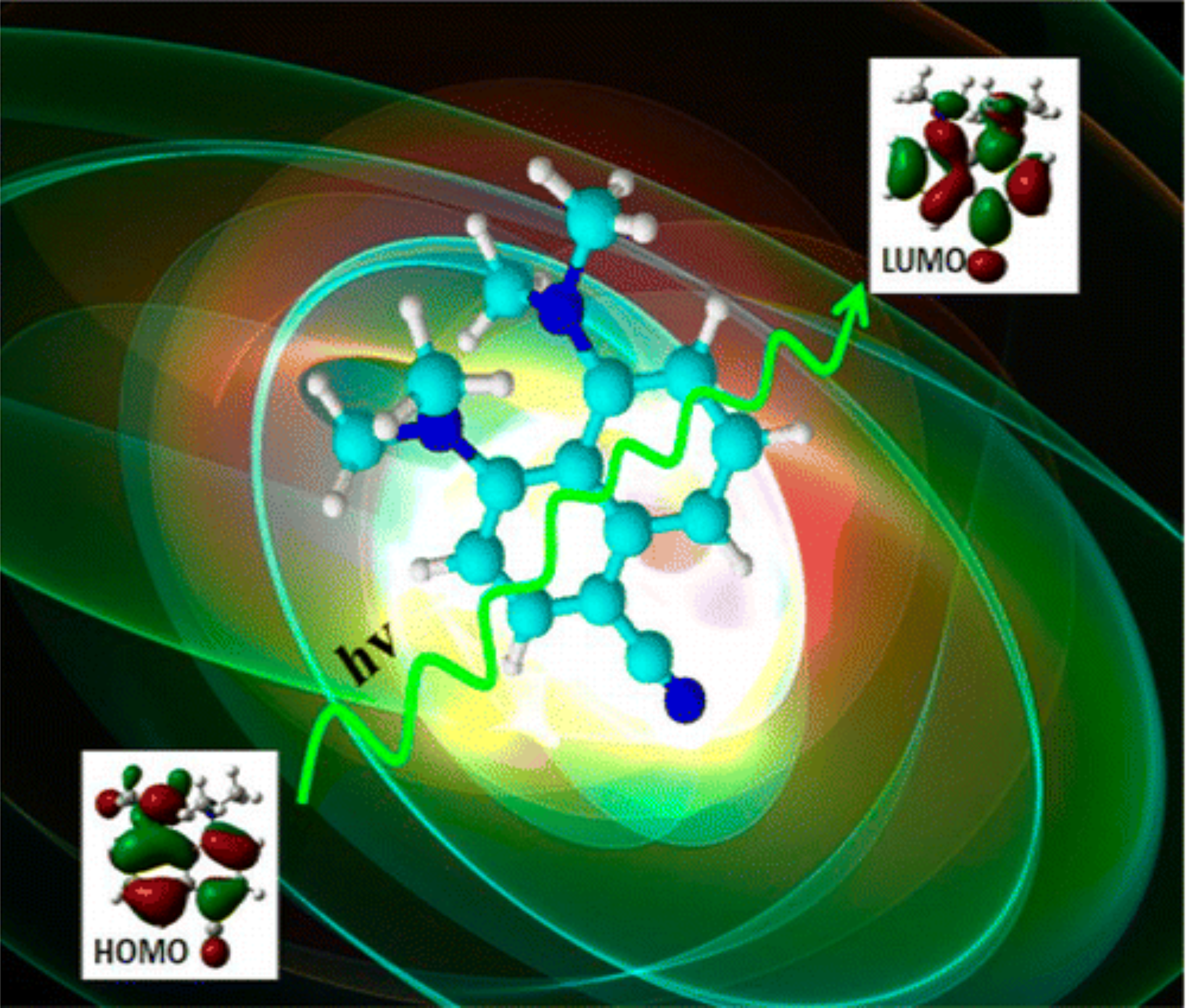
Ultrafast Excited-State Dynamics of a Cyano-Substituted "Proton Sponge"
G. Balkowski, A. Szemik-Hojniak, W. J. Buma, H. Zhang, I. Deperasinska, L. Poisson, J. M. Mestdagh, G. Piani and K. Oberda
J. Phys. Chem. A, 119, 11233, 2015 [doi]
The dynamics of a substituted proton sponge the 1,8-bis(dimethylamino)-4-cyanonaphthalene (DMAN-CN) molecule was investigated after excitation in the S-1 state. Experimental and theoretical information are reported. The former includes absorption, flucirescence, and time-resolved transient absorption spectra, which 'were recorded in -solution. Real-time dynamics measurements were also performed on gas-phase isolated DMAN-CN. TD DFT/6-31G(d,p) level and CIS/6-31G(d,p) excited-state calculations complement these results. This has allowed revisiting the energy transfer process between a locally excited (LE) and a charge transfer (CT) state, which is often invoked with this kind of molecule.

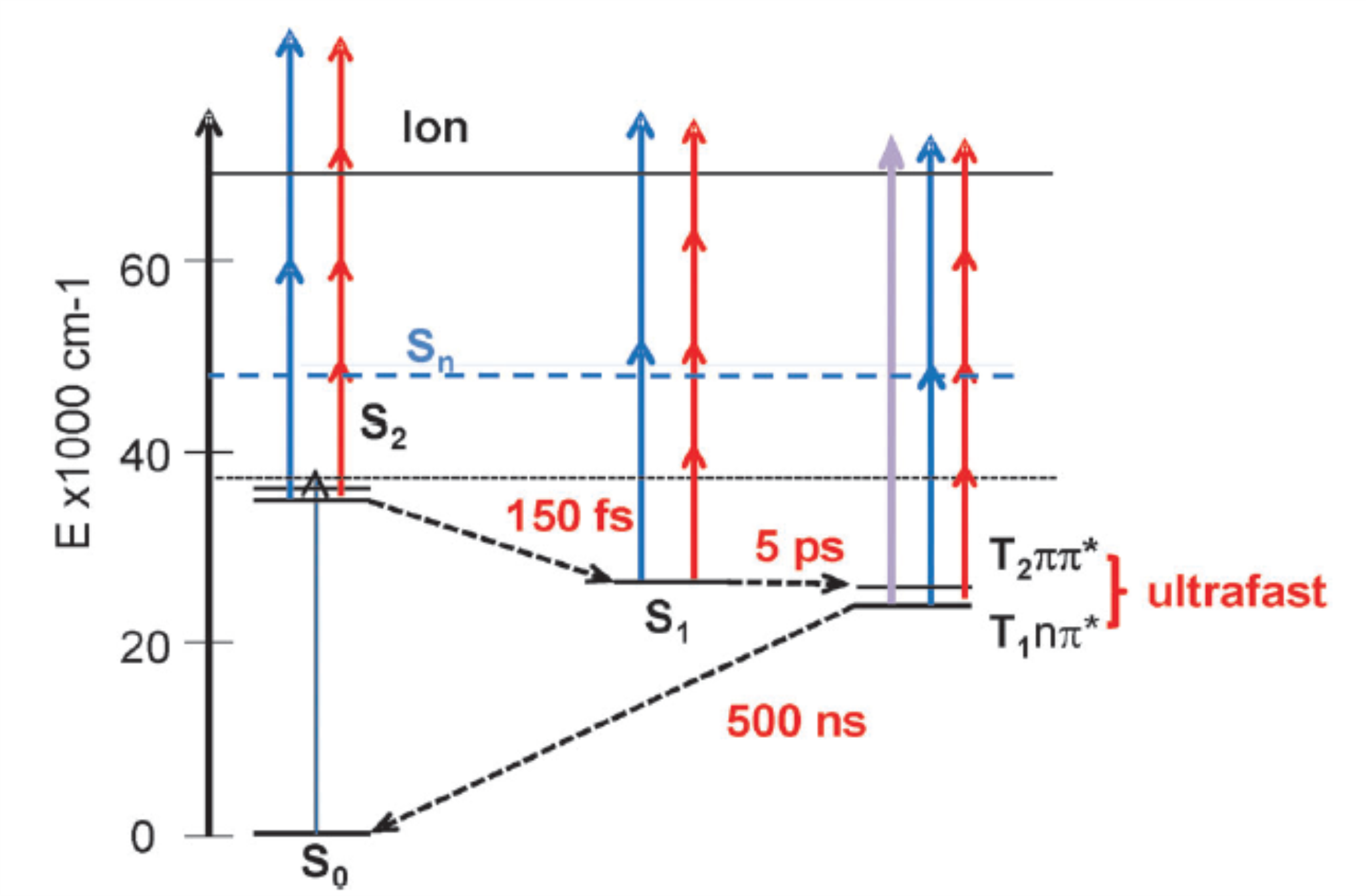
Direct observation of slow intersystem crossing in an aromatic ketone, fluorenone
B. Soep, J. M. Mestdagh, M. Briant and M. A. Gaveau
Phys. Chem. Chem. Phys., 18, 22914, 2016 [doi]
Direct measurements of Single vibronic Level InterSystem Crossing (SLISC) have been performed on the fluorenone molecule in the gas phase, by time resolved photoelectron and photoion spectroscopy. Vibronic transitions above the S-1 n pi* origin were excited in the 432-420 nm region and the decay of S-1 and growth of T-1 (3) pi pi* could be observed within a 10 ns time domain. The ionization potential is measured as 8.33 +/- 0.04 eV. The energy of the first excited triplet state of fluorenone, T-1 has been characterized directly at 18640 +/- 250 cm(-1). The internal conversion of S-1 to S-0 is found to amount to similar to 15% of the population decay, thus ISC is the dominant electronic relaxation process. ISC, although favored by the S-1 (1)n pi*-T-1 3 pi pi* coupling scheme, is 3 orders of magnitude less efficient than in the similar molecule benzophenone. Thus, the planarity of the fluorenone molecule disfavors the exploration of the configuration space where surface crossings would create high ISC probability, which occurs in benzophenone through surface crossings. The time evolution of S-1 fluorenone is well accounted for by the statistical decay of individual levels into a quasi-continuum of T-1 vibronic levels.
• ![]() Laser-matter interaction › Physico-chemistry and Chemical-physics
Laser-matter interaction › Physico-chemistry and Chemical-physics
• ![]() Institut Rayonnement Matière de Saclay
Institut Rayonnement Matière de Saclay
• LUMO-DyR - Reaction Dynamics Team • LUMO - Equipe Dynamique Réactionnelle (DyR)
