










Photoionization of 2-pyridone and 2-hydroxypyridine
J. C. Poully, J. P. Schermann, N. Nieuwjaer, F. Lecomte, G. Gregoire, C. Desfrancois, G. A. Garcia, L. Nahon, D. Nandi, L. Poisson and M. Hochlaf
Phys. Chem. Chem. Phys., 12, 3566, 2010.
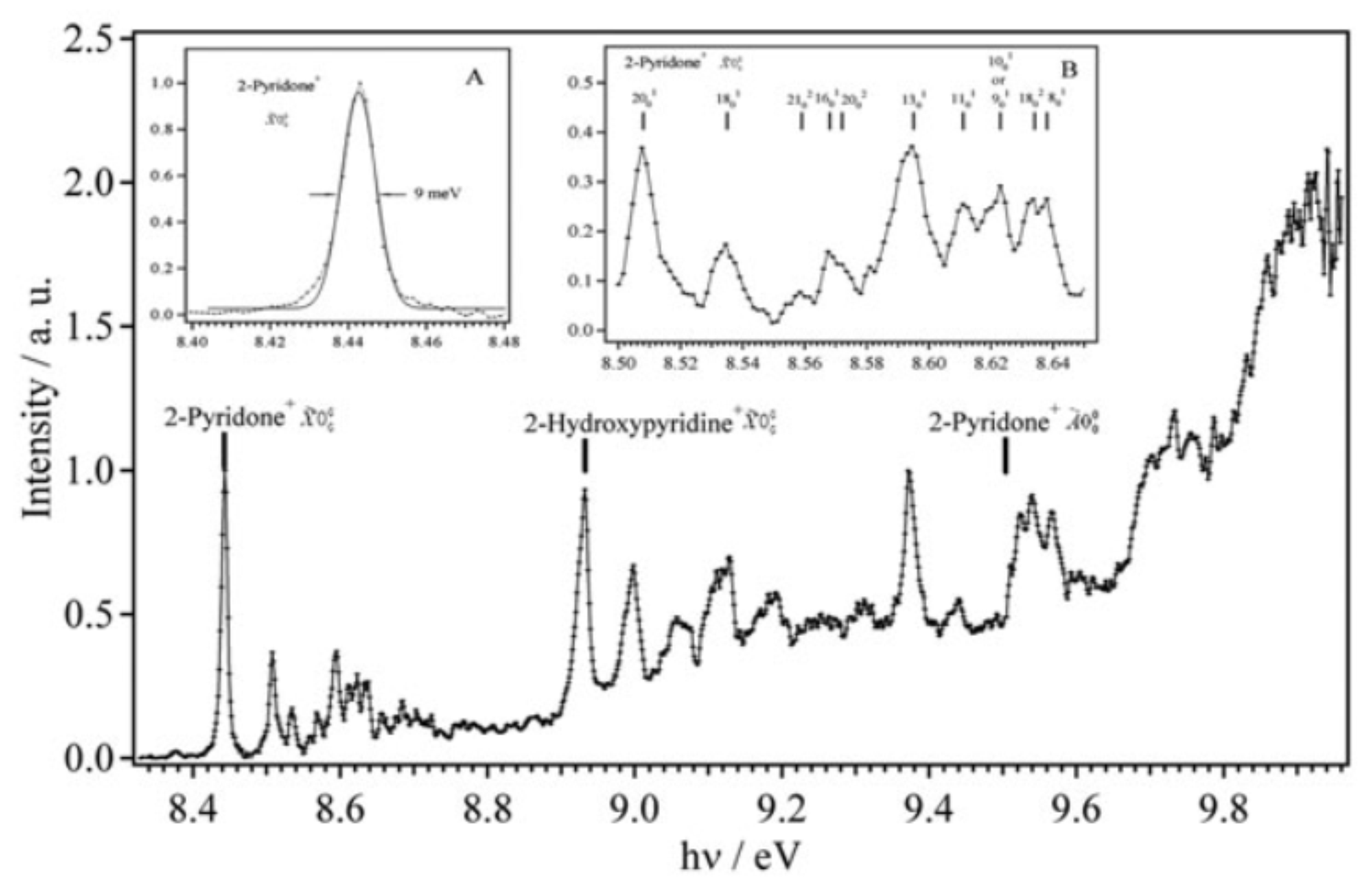
We studied the photoionization of 2-pyridone and its tautomer, 2-hydroxypyridine by means of VUV synchrotron radiation coupled to a velocity map imaging electron/ion coincidence spectrometer. The photoionization efficiency (PIE) spectrum is composed of steps. The state energies of the [2-pyridone]+ cation in the [X with combining tilde] ground and à excited electronic states, as well as of the [2-hydroxypyridine]+ cation in the electronic ground state, are determined. The slow photoelectron spectra (SPES) are dominated by the 000 transitions to the corresponding electronic states together with several weaker bands corresponding to the population of the pure or combination vibrational bands of the cations. These vibrationally-resolved spectra compare very well with state-of-the-art calculations. Close to the ionization thresholds, the photoionization of these molecules is found to be mainly dominated by a direct process whereas the indirect route (autoionization) may contribute at higher energies.

State-Selected Unimolecular Decomposition of δ-Valerolactam+ and δ-Valerolactam2+ Cations: Theory and Experiment
A. Mahjoub, M. Hochlaf, G. A. Garcia, L. Nahon and L. Poisson
J. Phys. Chem. A, 116, 8706, 2012.
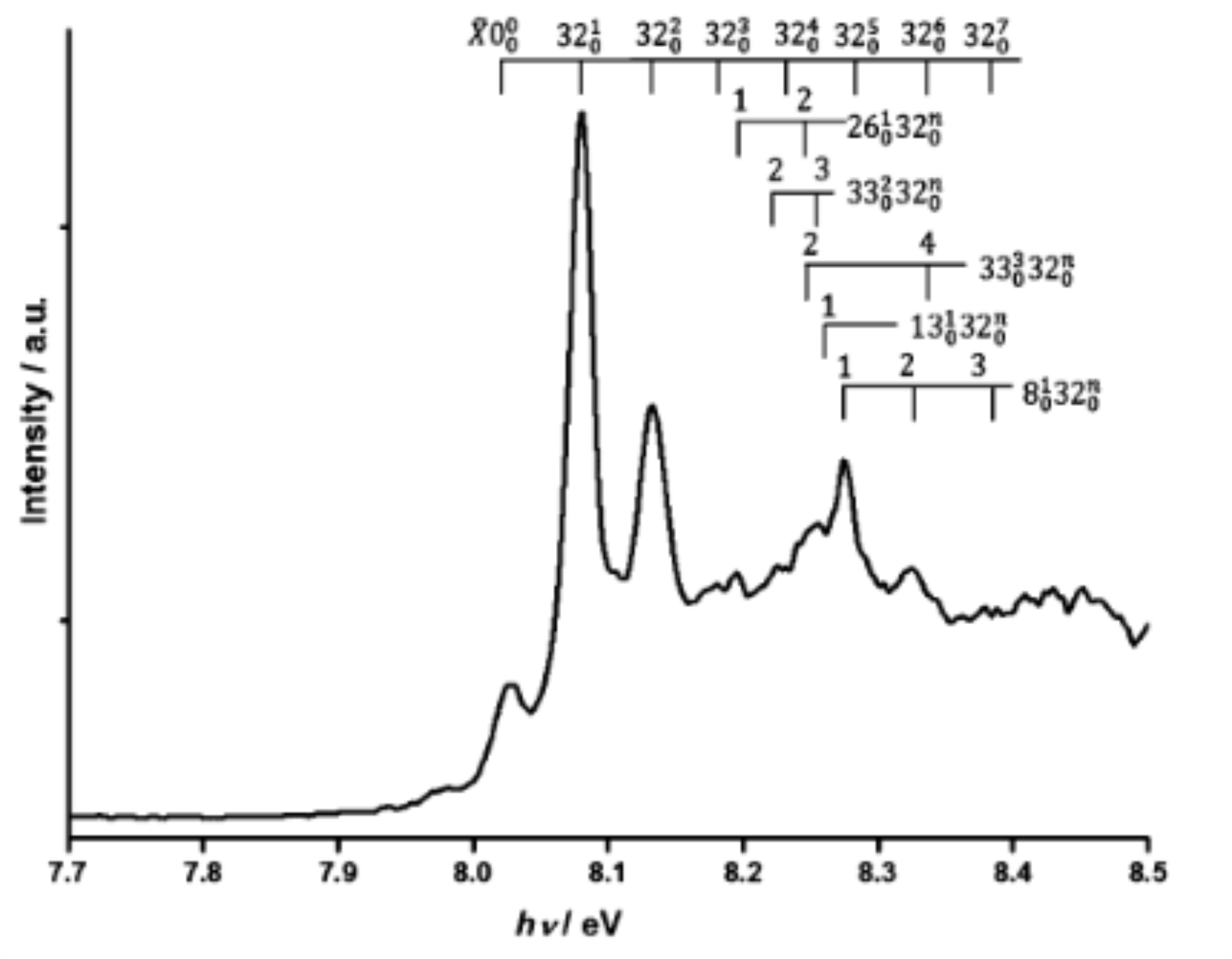
The near threshold photofragmentation pattern of δ-valerolactam+ and δ-valerolactam2+ has been recorded combining electron/ion coincidence techniques and vacuum ultraviolet synchrotron radiation. The experimental method yields the fragment intensity as a function of the internal energy deposited into the parent cation, up to 3.1 eV above the first ionization threshold. In parallel, ab initio studies on the δ-valerolactam+ and δ-valerolactam2+ cations and their ionic and neutral fragmentation products have been performed with the aim of determining the isomers of the ionic products observed experimentally as well as of their neutral counterparts. These computations were performed using the PBE0 exchange-correlation functional and the aug-cc-pVDZ basis set. We found good agreement between the calculated reaction enthalpies and experimental appearance energies of the ions. More generally, our experimental and theoretical results reveal that the fragmentation of the ionic species of interest leads to a multitude of neutral and ionic fragments, which may be formed after intramolecular isomerization and complex decomposition processes. Multistep reaction pathways are expected.
Slow Photoelectron Spectroscopy of 3-Hydroxyisoquinoline
Y. Pan, K.-C. Lau, L. Poisson, G. A. Garcia, L. Nahon and M. Hochlaf
J. Phys. Chem. A, 117, 8095, 2013.
We studied the single photon ionization of gas phase 3-hydroxyisoquinoline by means of VUV synchrotron radiation coupled to a velocity map imaging electron/ion coincidence spectrometer. Near the ionization thresholds of 3-hydroxyisoquinoline, the photoionization is found to occur mainly via a direct process. The spectra are assigned with the help of theoretical calculations on the equilibrium geometries, electronic states patterns, harmonic and anharmonic wavenumbers of the lactim and lactam forms of 3-hydroxyisoquinoline and their cations. The slow photoelectron spectrum (SPES) of this lactim is dominated by vibrational transitions to the X̃ state of the cation. In addition, several weaker and complex bands are observed, corresponding to the population of the vibrational bands (pure or combination) of the à electronically excited state of the cation. The adiabatic ionization energy of 3-hydroxyisoquinoline and the lowest electronic state energetics of the lactim and lactam cationic forms are determined.
Vibrationally Resolved Photoelectron Spectroscopy of Electronic Excited States of DNA Bases: Application to the à State of Thymine Cation
M. Hochlaf, Y. Pan, K.-C. Lau, Y. Majdi, L. Poisson, G. A. Garcia, L. Nahon, M. M. Al Mogren and M. Schwell
J. Phys. Chem. A, 119, 1146, 2015.
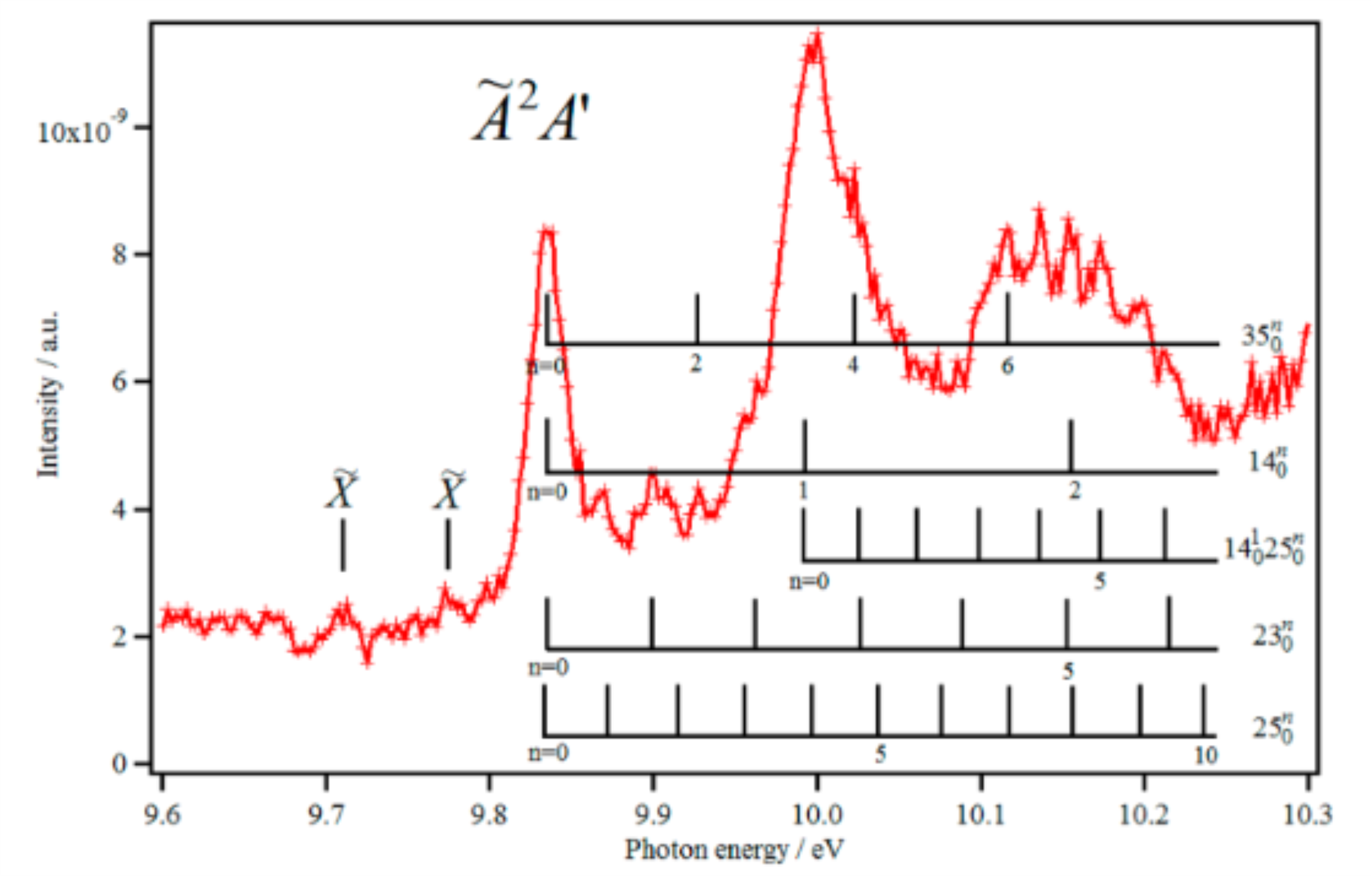
For fully understanding the light–molecule interaction dynamics at short time scales, recent theoretical and experimental studies proved the importance of accurate characterizations not only of the ground (D0) but also of the electronic excited states (e.g., D1) of molecules. While ground state investigations are currently straightforward, those of electronic excited states are not. Here, we characterized the à electronic state of ionic thymine (T+) DNA base using explicitly correlated coupled cluster ab initio methods and state-of-the-art synchrotron-based electron/ion coincidence techniques. The experimental spectrum is composed of rich and long vibrational progressions corresponding to the population of the low frequency modes of T+(Ã). This work challenges previous numerous works carried out on DNA bases using common synchrotron and VUV-based photoelectron spectroscopies. We provide hence a powerful theoretical and experimental framework to study the electronic structure of ionized DNA bases that could be generalized to other medium-sized biologically relevant systems.
Theoretical and Experimental Photoelectron Spectroscopy Characterization of the Ground State of Thymine Cation
Y. Majdi, M. Hochlaf, Y. Pan, K. C. Lau, L. Poisson, G. A. Garcia, L. Nahon, M. M. Al-Mogren and M. Schwell
J. Phys. Chem. A, 119, 5951, 2015
We report on the vibronic structure of the ground state (X) over tilde (2)A '' of the thymine cation, which has been measured using a threshold photoelectron photoion coincidence technique and vacuum ultraviolet synchrotron radiation. The threshold photoelectron spectrum, recorded over similar to 0.7 eV above the ionization potential (i.e., covering the whole ground state of the cation) shows rich vibrational structure that has been assigned with the help of calculated anharmonic modes of the ground electronic cation state at the PBE0/aug-cc-pVDZ level of theory. The adiabatic ionization energy has been experimentally determined as AIE = 8.913 +/- 0.005 eV, in very good agreement with previous high resolution results. The corresponding theoretical value of AIE = 8.917 eV has been calculated in this work with the explicitly correlated method/basis set (R)CCSD(T)-F12/cc-pVTZ-F12, which validates the theoretical approach and benchmarks its accuracy for future studies of medium-sized biological molecules.

Identifying Cytosine-Specific Isomers via High-Accuracy Single Photon Ionization
Z. Chen, K.-C. Lau, G. A. Garcia, L. Nahon, D. K. Božanić, L. Poisson, M. M. Al-Mogren, M. Schwell, J. S. Francisco, A. Bellili and M. Hochlaf
J. Am. Chem. Soc, 138, 16596, 2016.
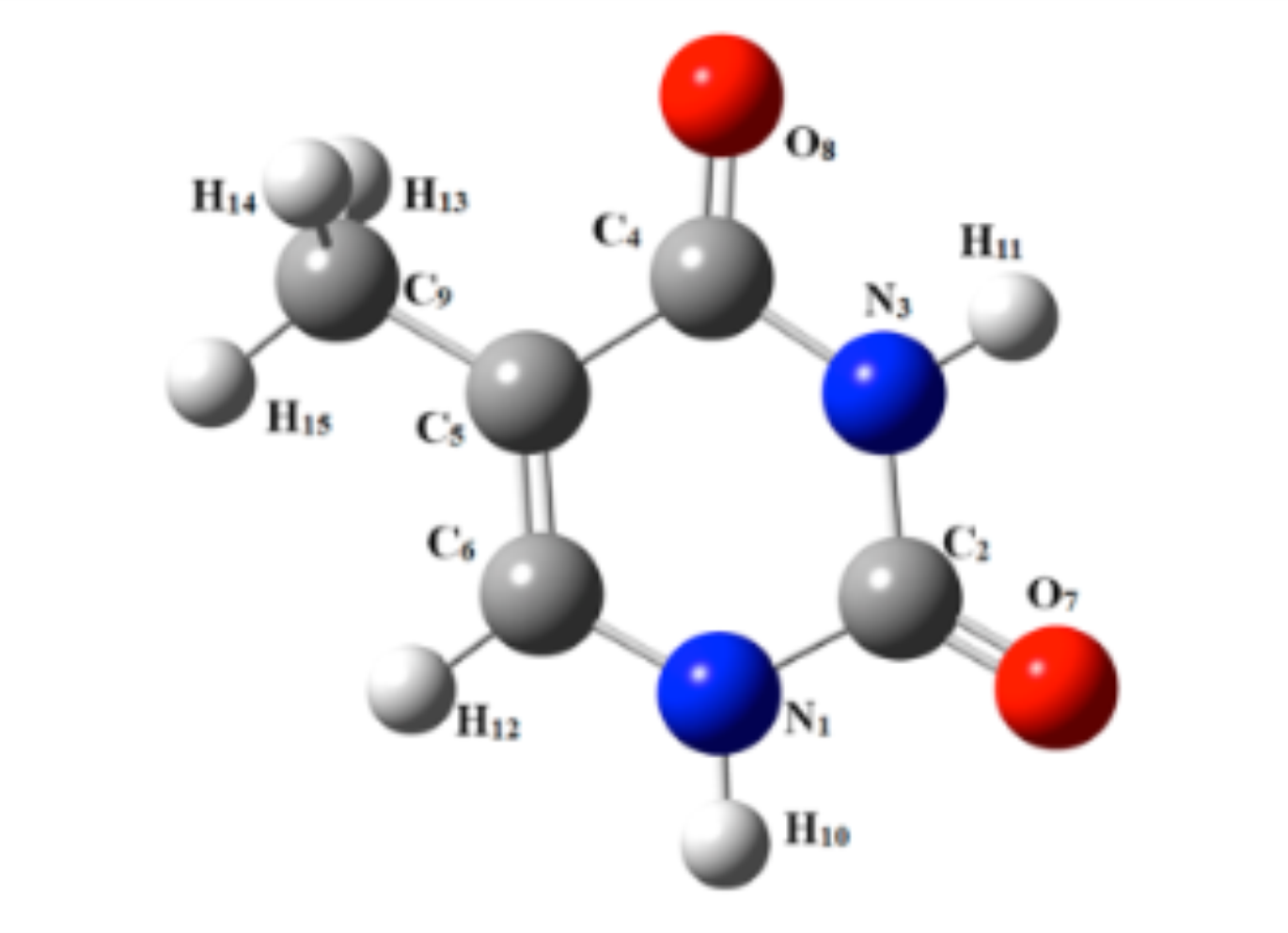
Biological entities, such as DNA bases or proteins, possess numerous tautomers and isomers that lie close in energy, making the experimental characterization of a unique tautomer challenging. We apply VUV synchrotron-based experiments combined with state-of-the-art ab initio methodology to determine the adiabatic ionization energies (AIEs) of specific gas-phase cytosine tautomers produced in a molecular beam. The structures and energetics of neutral and cationic cytosine tautomers were determined using explicitly correlated methods. The experimental spectra correspond to well-resolved bands that are attributable to the specific contributions of five neutral tautomers of cytosine prior to ionization. Their AIEs are experimentally determined for the first time with an accuracy of 0.003 eV. This study also serves as an important showcase for other biological entities presenting a dense pattern of isomeric and tautomeric forms in their spectra that can be investigated to understand the charge redistribution in these species upon ionization.
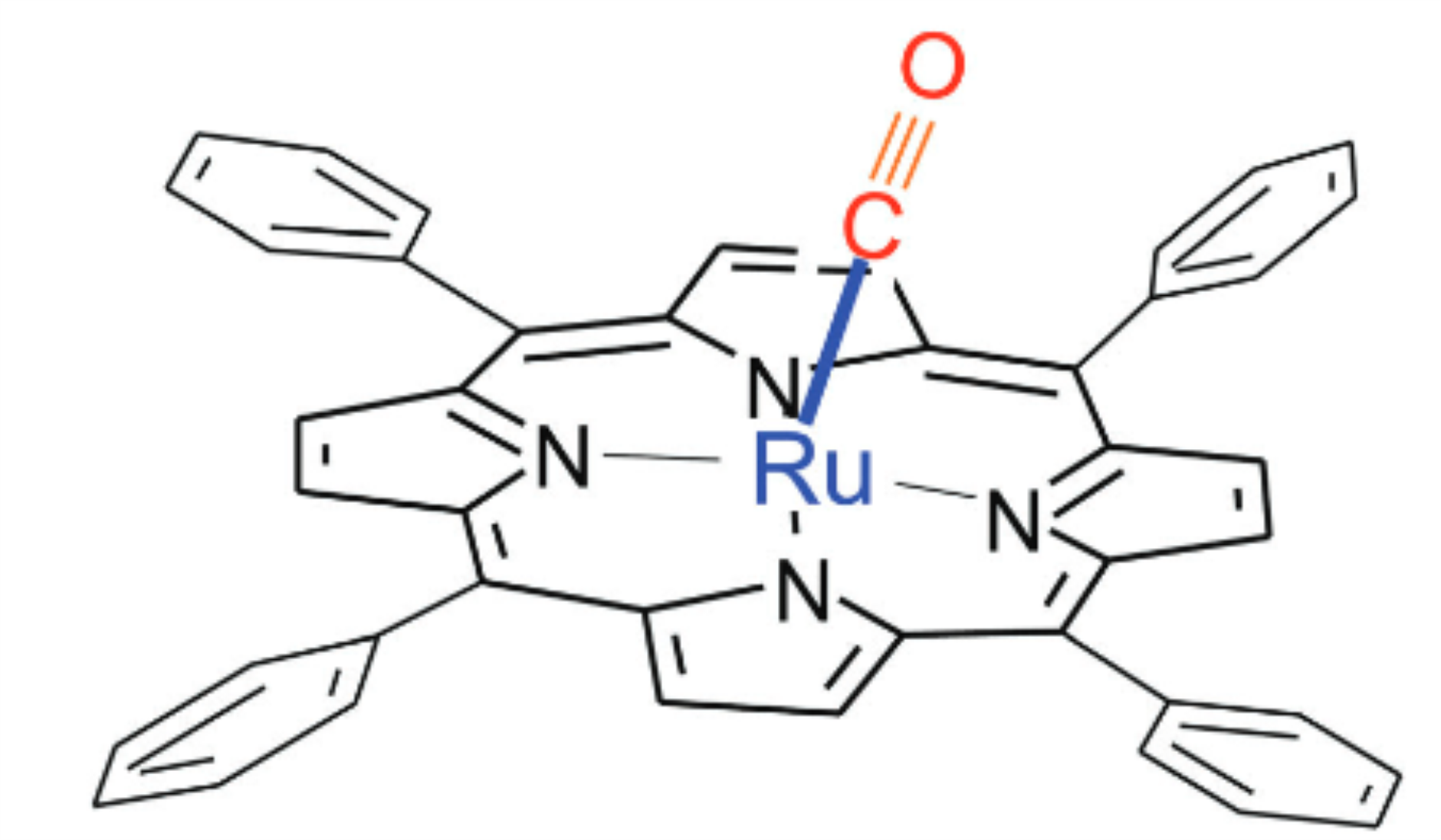
The surprisingly high ligation energy of CO to ruthenium porphyrins
N. Shafizadeh, S. Boyé-Péronne, S. Soorkia, B. K. Cunha de Miranda, G. A. Garcia, L. Nahon, S. Chen, A. de la Lande, L. Poisson and B. Soep
Phys. Chem. Chem. Phys., 20, 11730, 2018.
A combined theoretical and experimental approach has been used to investigate the binding energy of a ruthenium metalloporphyrin ligated with CO, ruthenium tetraphenylporphyrin [RuII TPP], in the RuII oxidation degree. Measurements performed with VUV ionization using the DESIRS beamline at Synchrotron SOLEIL led to adiabatic ionization energies of [RuII TPP] and its complex with CO, [RuII TPP–CO], of 6.48 ± 0.03 eV and 6.60 ± 0.03 eV, respectively, while the ion dissociation threshold of [RuII TPP–CO]+ is measured to be 8.36 ± 0.03 eV using the ground-state neutral complex. These experimental data are used to derive the binding energies of the CO ligand in neutral and cationic complexes (1.88 ± 0.06 eV and 1.76 ± 0.06 eV, respectively) using a Born–Haber cycle. Density functional theory calculations, in very satisfactory agreement with the experimental results, help to get insights into the metal–ligand bond. Notably, the high ligation energies can be rationalized in terms of the ruthenium orbital structure, which is singular compared to that of the iron atom. Thus, beyond indications of a strengthening of the Ru–CO bond due to the decrease in the CO vibrational frequency in the complex as compared to the Fe–CO bond, high-level calculations are essential to accurately describe the metal ligand (CO) bond and show that the Ru–CO bond energy is strongly affected by the splitting of triplet and singlet spin states in uncomplexed [Ru TPP].

Unveiling the complex vibronic structure of the canonical adenine cation
H. Y. Zhao, K.-C. Lau, G. A. Garcia, L. Nahon, S. Carniato, L. Poisson, M. Schwell, M. M. Al-Mogren and M. Hochlaf
Phys. Chem. Chem. Phys., 20, 20756, 2018.
Adenine, a DNA base, exists as several tautomers and isomers that are closely lying in energy and that may form a mixture upon vaporization of solid adenine. Indeed, it is challenging to bring adenine into the gas phase, especially as a unique tautomer. The experimental conditions were tuned to prepare a jet-cooled canonical adenine (9H-adenine). This isolated DNA base was ionized by single VUV photons from a synchrotron beamline and the corresponding slow photoelectron spectrum was compared to ab initio computations of the neutral and ionic species. We report the vibronic structure of the X+ 2A′′ (D0), A+ 2A′ (D1) and B+ 2A′′ (D2) electronic states of the 9H adenine cation, from the adiabatic ionization energy (AIE) up to AIE + 1.8 eV. Accurate AIEs are derived for the 9H-adenine (̃ 1A′) + hν → 9H-adenine+ (X+ 2A′′, A+ 2A′, B+ 2A′′) + e− transitions. Close to the AIE, we fully assign the rich vibronic structure solely to the 9H-adenine (X 1A′) + hν → 9H-adenine+ (X+ 2A′′) transition. Importantly, we show that the lowest cationic electronic states of canonical adenine are coupled vibronically. The present findings are important for understanding the effects of ionizing radiation and the charge distribution on this elementary building block of life, at ultrafast
• ![]() Interaction laser-matière › Physico-Chimie et Chimie-Physique
Interaction laser-matière › Physico-Chimie et Chimie-Physique
• Laboratoire Interactions, Dynamiques et Lasers (LIDYL) - CEA-CNRS et Université Paris Saclay
• LUMO-DyR - Reaction Dynamics Team • LUMO - Equipe Dynamique Réactionnelle (DyR)
