









Femtosecond dynamics of the tert-butyl radical, t-C4H9
B. Noller, R. Maksimenka, I. Fischer, M. Armone, B. Engels, C. Alcaraz, L. Poisson and J. M. Mestdagh
J. Phys. Chem. A, 111, 1771, 2007 [doi]
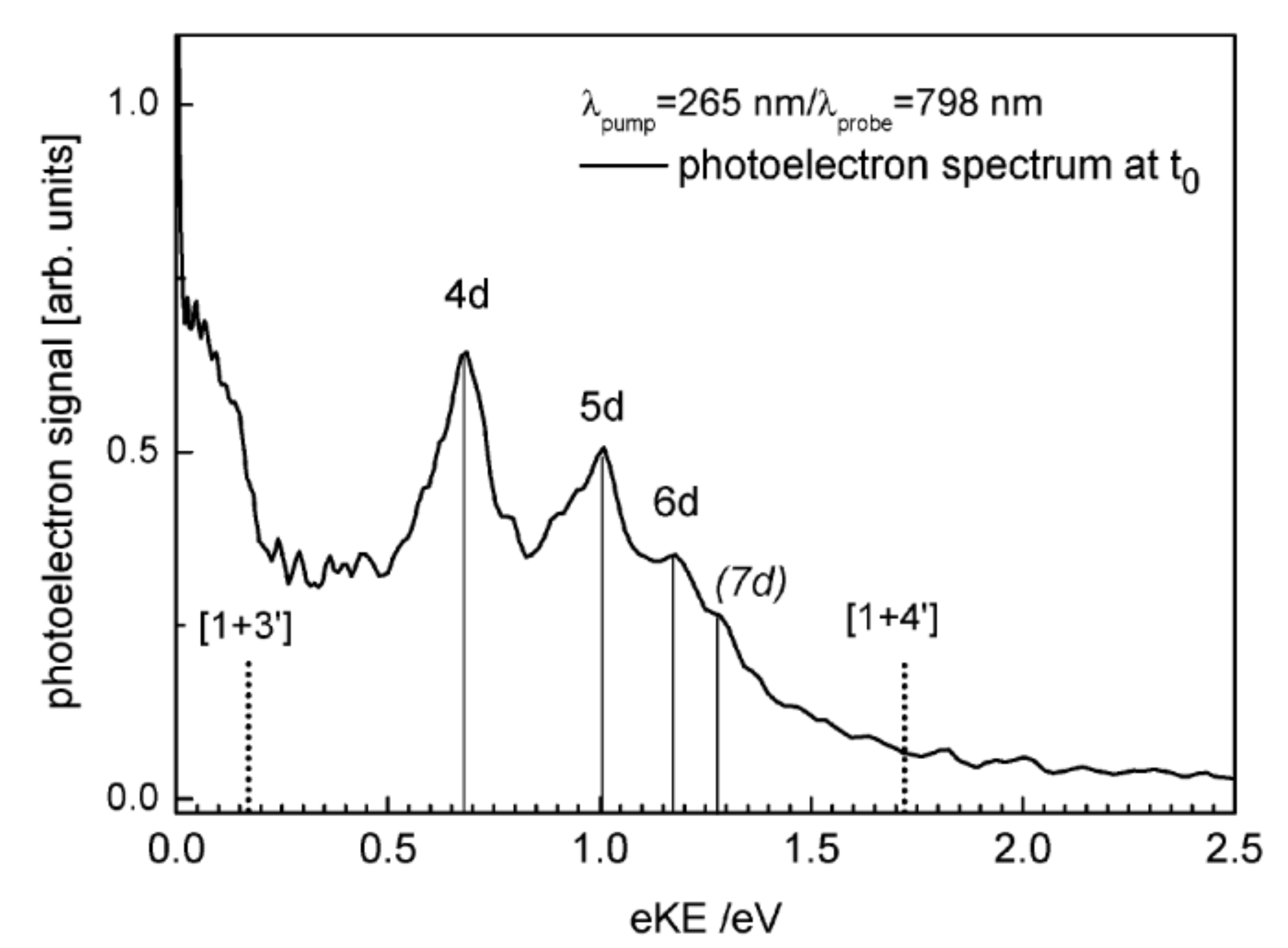
The excited-state dynamics of the tert-butyl radical, t-C4H9, was investigated by femtosecond time-resolved photoionization and photoelectron spectroscopy. The experiments were supported by ab initio calculations. tert-Butyl radicals, generated by flash pyrolysis of azo-tert-butane, were excited into the A (2)A(1) (3s) state between 347 and 307 nm and the 3p band at 274 and 268 nm and ionized by 810-nm radiation, in a [1 + 2'] or [1 + 3'] process. Electronic structure calculations confirm that the two states are of s and p Rydberg characters, respectively. The carbon framework becomes planar and thus ion-like in both states. The photoelectron spectra are broad and seem to be mediated by accidental intermediate resonances in the probe step. All time-resolved photoelectron spectra can be described by a single decay time. For the A (2)A(1) state, lifetimes between 180 and 69 fs were measured. Surprisingly, a much longer lifetime of around 2 ps was found for the 3p state. To understand the decay dynamics, the potential energy was computed as a function of several important nuclear coordinates. A [1,2] H-atom shift to the isobutyl radical seems not to be important for the excited-state dynamics. Qualitative considerations indicate curve crossings between the ground state, the 3s state, and a valence state along the asymmetric C-C stretch coordinate that correlates to the dimethylcarbene + methyl product channel. The implications of the present study for earlier work on the nanosecond time scale are discussed.

Femtosecond Dynamics of Isolated Phenylcarbenes
B. Noller, L. Poisson, R. Maksimenka, I. Fischer and J. M. Mestdagh
J. Am. Chem. Soc, 130, 14908, 2008 [doi]
Understanding the primary photophysical processes in molecules is essential for interpreting their photochemistry, because molecules rarely react from the initially excited electronic state. In this study the ultrafast excited-state dynamics of chlorophenylcarbene (CPC) and trifluoromethylphenylcarbene (TFPC), two species that are considered as models for carbene dynamics, were investigated by femtosecond time-resolved pump probe spectroscopy in the gas phase. Their dynamics was followed in real time by time-resolved photoionization and photoelectron imaging. CPC was excited at 265 nm into the 3 (1)A' state, corresponding to excitation from a TT-orbital of the aromatic ring into the LUMO. The LUMO contains a contribution of the p-orbital at the carbene center. Three time constants are apparent in the photoelectron images: A fast decay process with T-1 approximate to 40 fs, a second time constant of T-2 approximate to 350 fs, and an additional time constant of T-3 approximate to 1 ps. The third time constant is only visible in the time-dependence of low kinetic energy electrons. Due to the dense manifold of excited states between 3.9 and 5 eV, known from ab initio calculations, the recorded time-resolved electron images show broad and unstructured bands. A clear population transfer between the states thus can not directly be observed. The fast deactivation process is linked to either a population transfer between the strongly coupled excited states between 3.9 and 5.0 eV or the movement of the produced wave packet out of the Franck-Condon region. Since the third long time constant is only visible for photoelectrons at low kinetic energy, evidence is given that this time constant corresponds to the lifetime of the lowest excited A (1)A' state. The remaining time constant reflects a deactivation of the manifold of states in the range 3.9-5.0 eV down to the A (1)A' state.
Ultrafast Dynamics of Isolated Phenylcarbenes Followed by Femtosecond Time-Resolved Velocity Map Imaging
B. Noller, L. Poisson, R. Maksimenka, O. Gobert, I. Fischer and J. M. Mestdagh
J. Phys. Chem. A, 113, 3041, 2009 [doi]
The ultrafast dynamics of two carbene model systems, chlorophenylcarbene (CPC) and trifluoromethylphenylcarbene (TFPC), has been studied in a molecular beam. Velocity map imaging aids optimizing the pyrolysis conditions for a clean generation of reactive intermediates by supersonic jet flash pyrolysis. The dynamics was followed in real time by time-resolved mass spectroscopy and photoion and photoelectron imaging. CPC was excited at 265 nm into the 3 'A' state, corresponding to excitation from a pi-orbital of the aromatic ring into the lowest unoccupied molecular orbital, which contains the p-orbital at the carbene center. The experimental results suggest a three step deactivation process in agreement with computations. TFPC exhibits two absorption bands in the 36000 cm(-1) and 41000 cm(-1) range and gives rise to very similar dynamics, although it has a triplet ground state. The experimental data were augmented by Computations.
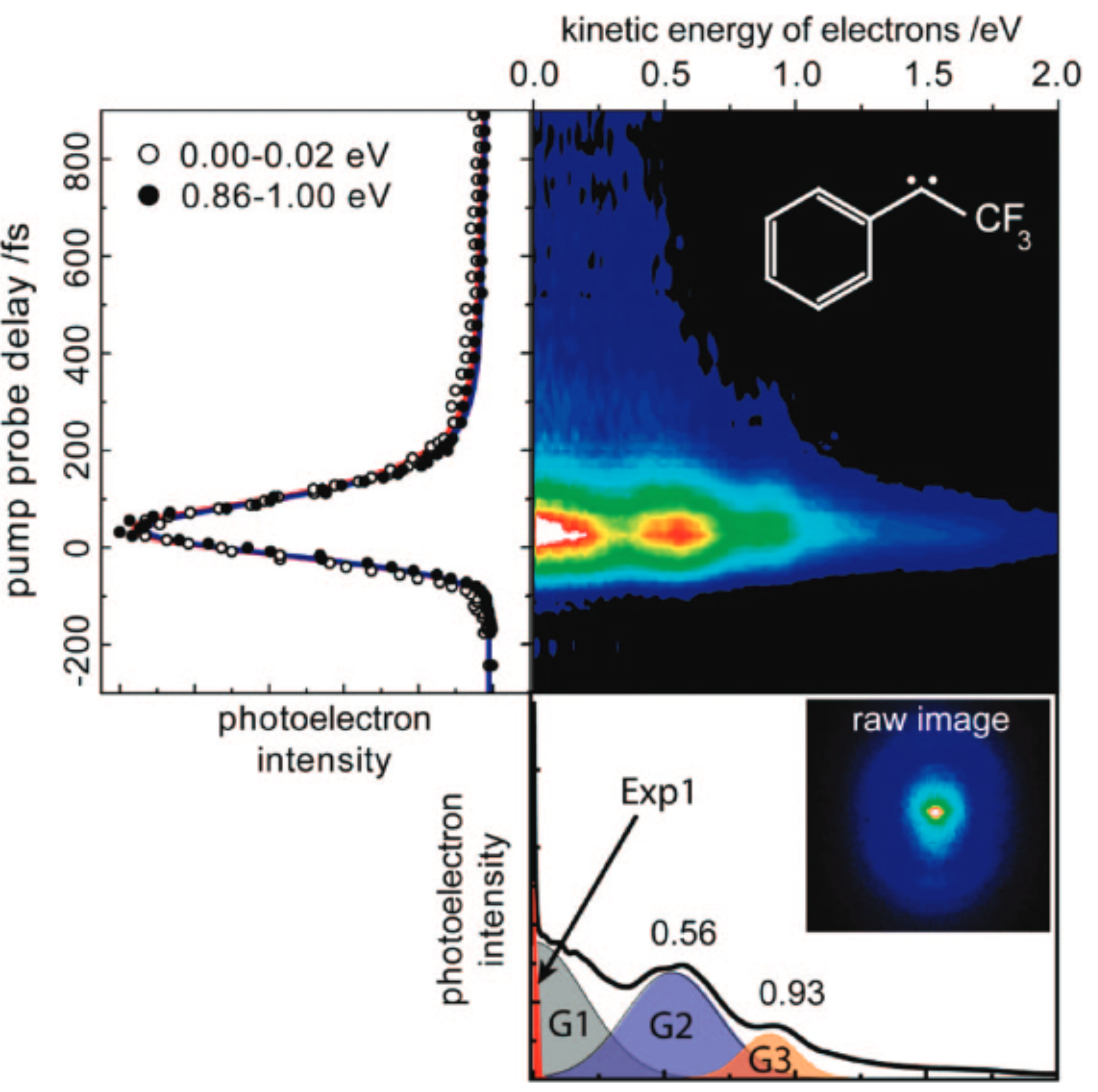
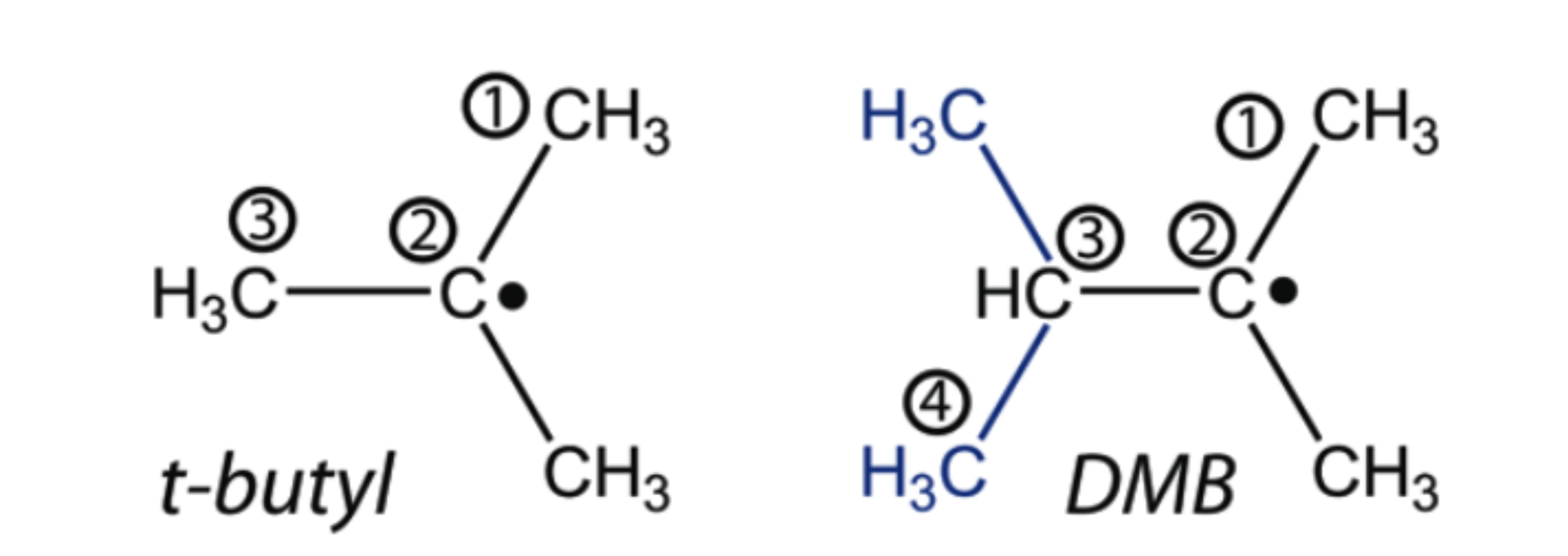
Side-Chain Effects on the Electronic Relaxation of Radicals followed by Time-Resolved Pump-Probe Spectroscopy: 2,3-Dimethylbut-2-yl vs tert-Butyl
B. Noller, L. Poisson, I. Fischer and J. M. Mestdagh
J. Phys. Chem. A, 114, 3045, 2010 [doi]
The excited-mate lifetime of the 2,3-dimethylbut-2-yl (DMB) radical, it hexyl isomer, upon electronic excitation into the 3p Rydberg state at 265 nm, is measured by femtosecond time-resolved photoionization. It is shown that the 3p state deactivates in I two-step process, which is well described by two mile constants of 25 and 400 fs The results tire compared to tert-butyl (t-C(4)H(9)), another tertiary radical investigated before. Time-dependent DFT calculations confirm the earlier suggestion that curve crossings along the C-C coordinate play all Important role in the excited-state deactivation
Femtosecond dynamics of cyclopropenylidene, c-C3H2
P. Hemberger, J. Koehler, I. Fischer, G. Piani, L. Poisson and J.-M. Mestdagh
Phys. Chem. Chem. Phys., 14, 6173, 2012 [doi]
The photophysics of the B B-1(1) state of isolated cyclopropenylidene, c-C3H2, has been studied by femtosecond time-resolved photoionisation and photoelectron spectroscopy. The carbene was produced by flash pyrolysis of 3-chlorocycloprop-1-ene. The bands at 266.9 nm and 264.6 nm have been investigated. The excited state deactivates in a two step process. The first time constant of less than 50 fs corresponds most likely to a nonradiative transition to the A-state, the second one on the order of 200 fs describes the internal conversion to the electronic ground state. The data are compared to those measured for the chlorinated carbene c-C3HCl. In the photoelectron spectrum of c-C3H2 resonances were observed which can be assigned to members of a Rydberg d-series.
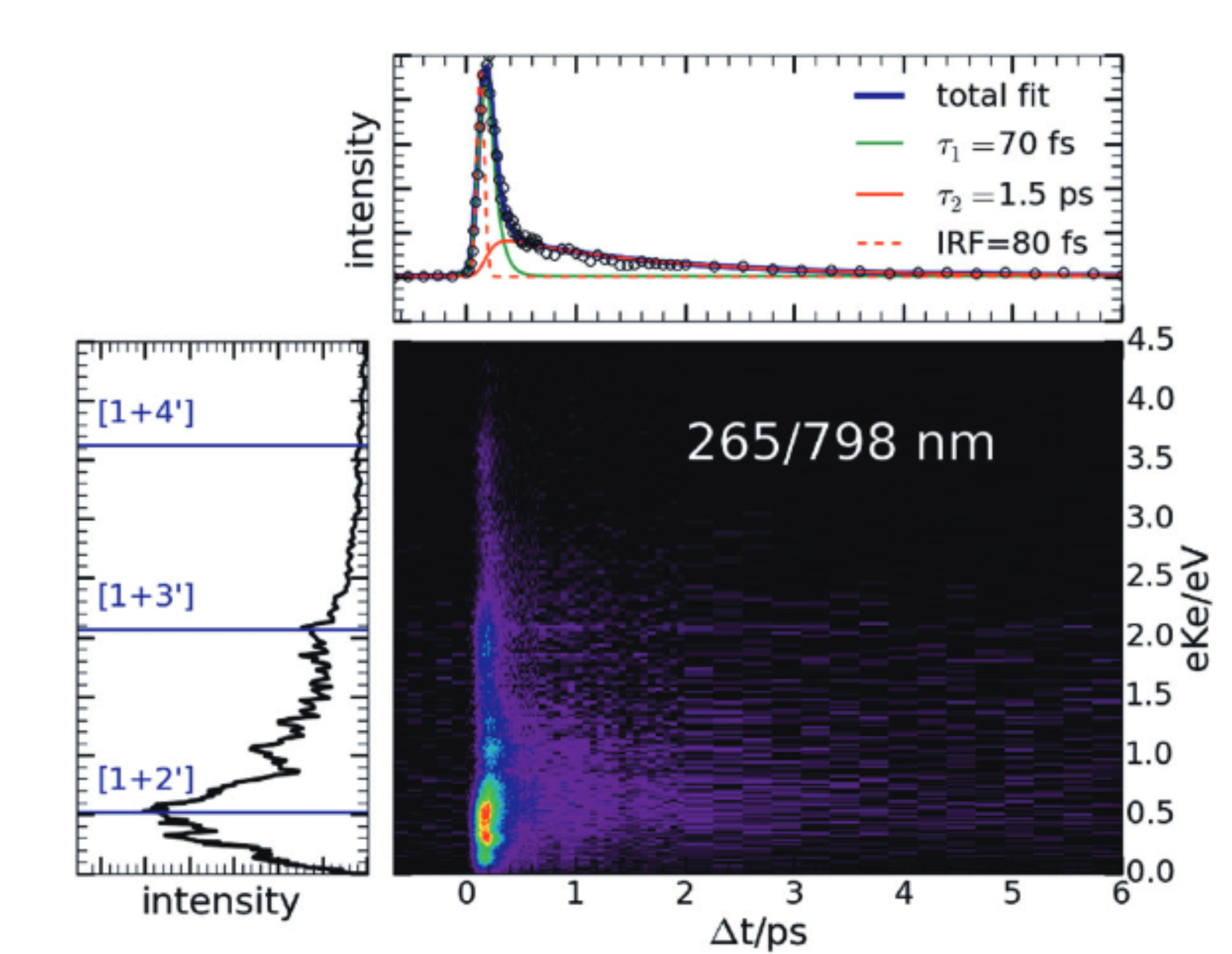
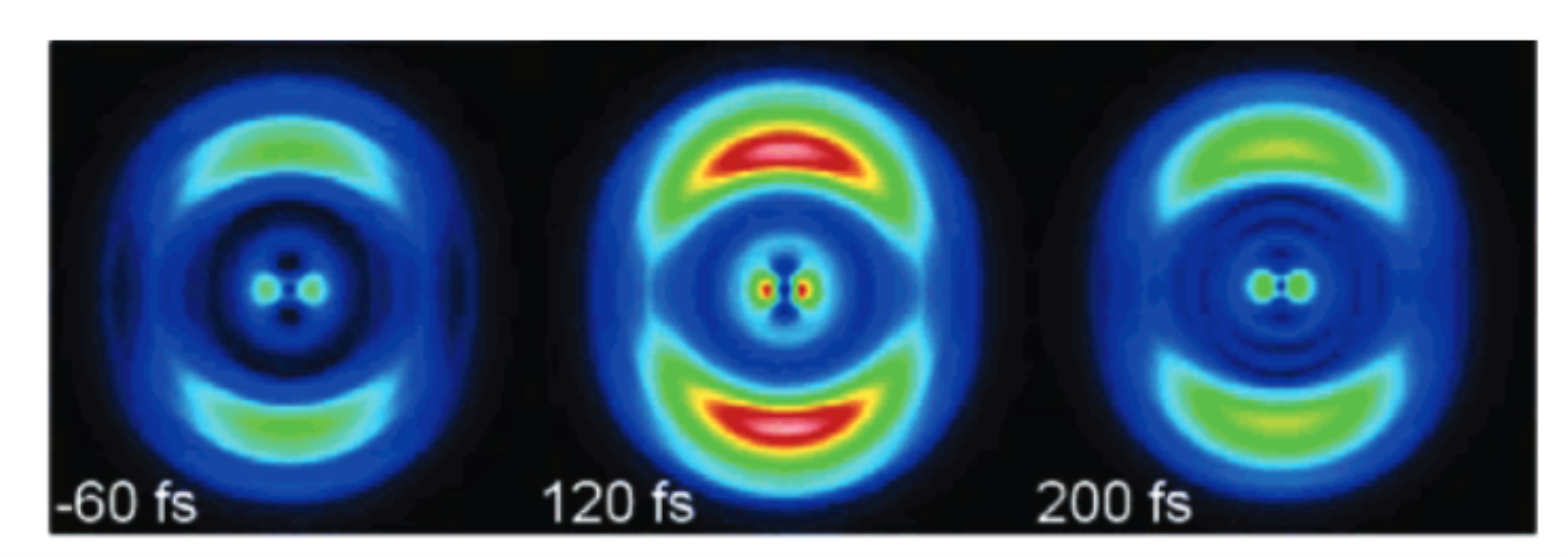
Femtosecond time-resolved photoelectron spectroscopy of the benzyl radical
A. Roder, A. Humeniuk, J. Giegerich, I. Fischer, L. Poisson and R. Mitric
Phys. Chem. Chem. Phys., 19, 12365, 2017 [doi]
We present a joint experimental and computational study of the nonradiative deactivation of the benzyl radical, C7H7, after UV excitation. Femtosecond time-resolved photoelectron imaging was applied to investigate the photodynamics of the radical. The experiments were accompanied by excited state dynamics simulations using surface hopping. Benzyl has been excited at 265 nm into the D-band (pi pi*) and the dynamics was probed using probe wavelengths of 398 nm or 798 nm. At a probe wavelength of 398 nm a single time constant of around 70-80 fs was observed. When the dynamics was probed at 798 nm, a second time constant tau(2) = 1.5 ps was visible, which can be attributed to further non-radiative deactivation to the lower-lying D-1/D-2 states.
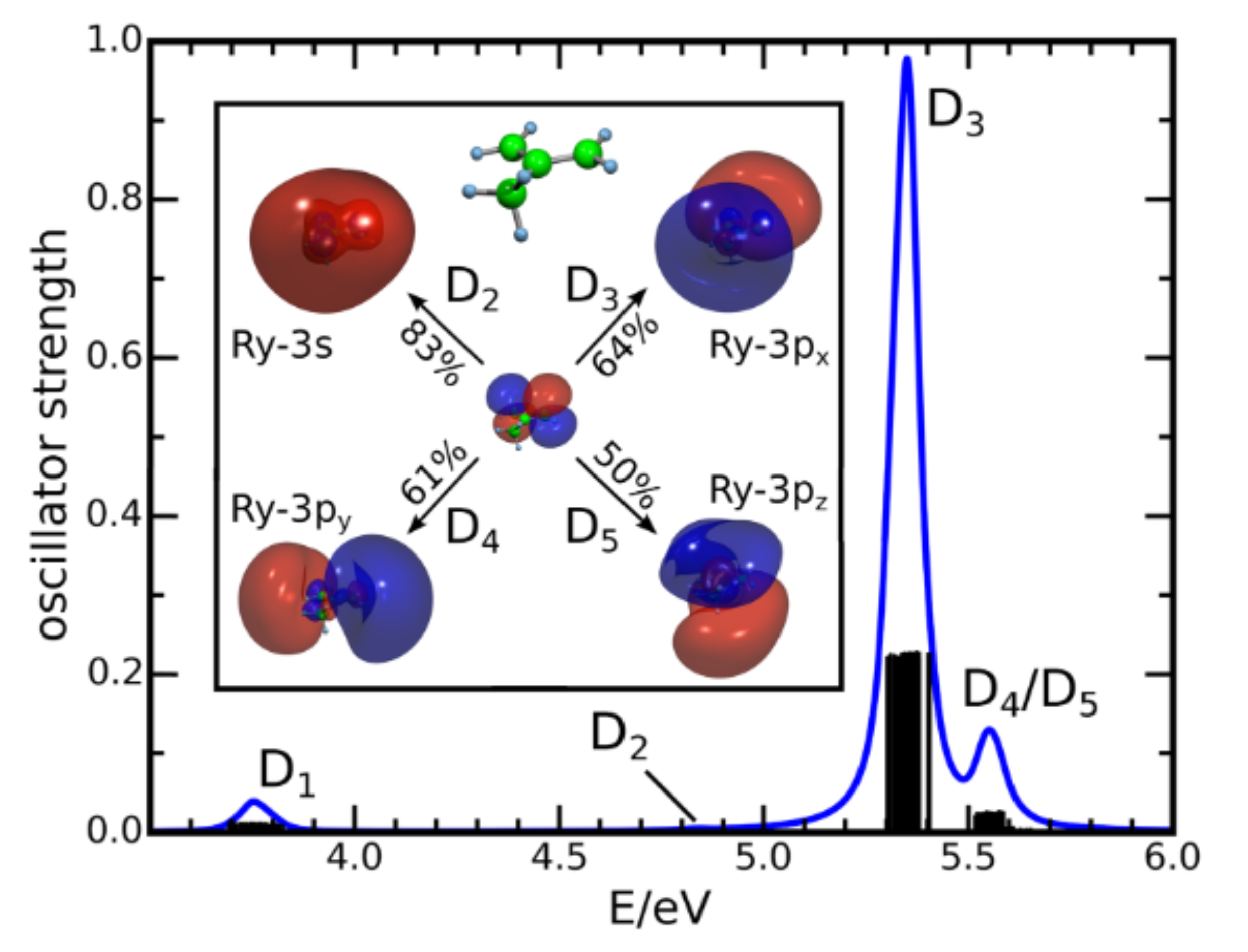
Femtosecond dynamics of the 2-methylallyl radical: A computational and experimental study
A. Roder, K. Issler, L. Poisson, A. Humeniuk, M. Wohlgemuth, M. Comte, F. Lepetit, I. Fischer, R. Mitric and J. Petersen
J. Chem. Phys., 147, 013902, 2017 [doi]
We investigate the photodynamics of the 2-methylallyl radical by femtosecond time-resolved photoelectron imaging. The experiments are accompanied by field-induced surface hopping dynamics calculations and the simulation of time-resolved photoelectron intensities and anisotropies, giving insight into the photochemistry and nonradiative relaxation of the radical. 2-methylallyl is excited at 236 nm, 238 nm, and 240.6 nm into a 3p Rydberg state, and the subsequent dynamics is probed by multiphoton ionization using photons of 800 nm. The photoelectron image exhibits a prominent band with considerable anisotropy, which is compatible with the result of theory. The simulations show that the initially excited 3p state is rapidly depopulated to a 3s Rydberg state, from which photoelectrons of high anisotropy are produced. The 3s state then decays within several 100 fs to the D-1 (n pi) state, followed by the deactivation of the D1 to the electronic ground state on the ps time scale. Published by AIP Publishing.

Excited state dynamics and time-resolved photoelectron spectroscopy of para-xylylene
K. Issler, A. Röder, F. Hirsch, L. Poisson, I. Fischer, R. Mitrić and J. Petersen
Faraday Discussions, 212, 83, 2018 [doi]
We investigated the excited-state dynamics of para-xylylene using a combination of field-induced surface hopping (FISH) simulations and time-resolved ionisation experiments. Our simulations predict an ultrafast decay of the initially excited bright state (S2/S3) to the S1 state on a sub-100 fs time scale, followed by return to the ground state within ∼1 ps. This is accompanied by a transient change of the biradical character of the molecule, as monitored by calculating natural orbital occupation numbers. Specifically, the initially low biradicality is increased by electronic excitation as well as by vibrational activation. Experimentally, para-xylylene was generated by pyrolysis from [2,2]paracyclophane and excited with 266 nm radiation into the S2/S3 bright state. The subsequent dynamics were followed using ionisation as the probe step, with both mass spectra and photoelectron spectra recorded as a function of pump–probe delay. The observed decay of photoelectron and photoion intensities closely matches the theoretical predictions and is consistent with the sequential mechanism found in the simulations. This mechanism exhibits characteristic signatures in both time-resolved mass and photoelectron spectra, in particular in the appearance of fragment ions that are exclusively generated from the S1 state. This allows for a separation of the S2 and S1 dynamics in the photoelectron and mass spectra. An excellent agreement between the observed and the simulated ion signal is observed.
