










|
Par comparaison avec des données expérimentales, spécifiquement obtenues dans ce but à l'IRAMIS/SPAM, il est possible de sélectionner la meilleure méthode ab initio, permettant de fixer les paramètres de modèles dits de "champ de force", pour reproduire fidèlement la structure d'assemblées d'atomes aussi complexe que celles constituant les protéines (macromolécule constituée d'acides aminés) ou les peptides (petits polymères d'acides aminés, n<50). |
La modélisation des protéines et des peptides joue un rôle majeur en biologie, notamment pour raffiner les structures des protéines ou simuler leur comportement dynamique. Afin de pouvoir prendre en compte de milliers d’atomes, ces modélisations nécessitent des descriptions simples du système à l’échelle moléculaire, qui s’appuient sur des modèles, dits de "champ de force", dépendants de paramètres en général déterminés par ajustements sur des calculs de chimie quantique.
Le choix des méthodes de chimie quantique employées est alors crucial, car il existe toute une palette de méthodes récentes permettant de traiter la corrélation électronique, phénomène à l’origine des forces de dispersion (forces de London) partout présentes dans les peptides et les protéines. Citons, outre les méthodes post-Hartree-Fock lourdes à mettre en œuvre, les méthodes issues de la Théorie de la Fonctionelle de la Densité, pures (DFT) ou comportant des paramètres de dispersion explicites (DFT-D).
Constatant l’absence de contreparties expérimentales directement comparables à ces calculs, c'est-à-dire réalisées en phase gazeuse, le groupe Structures Biomoléculaires du SPAM/LFP s’est lancé dans un programme (LASIHMODo : LASer investigations of Isolated and Hydrated peptides for a stepwise validation of advanced protein MODelling, financé par l’ANR) visant à étudier aussi finement que possible, par spectroscopie laser, des objets difficiles à modéliser.
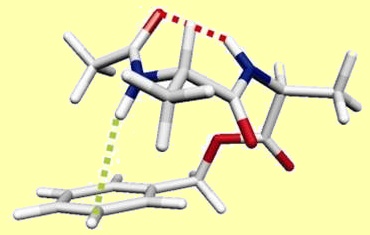
En relevant les défis de la vaporisation, par désorption laser, de l’identification des conformations présentes et de la mesure de leur abondance, par spectroscopie de double résonance IR/UV, il a été possible de montrer qu’un petit peptide modèle, l’acétyl-Ala-Ala-OBenzyl, choisi pour les difficultés théoriques qu’il soulève, comporte deux conformations stables à 300 K, l’une majoritaire étendue, l’autre minoritaire repliée.
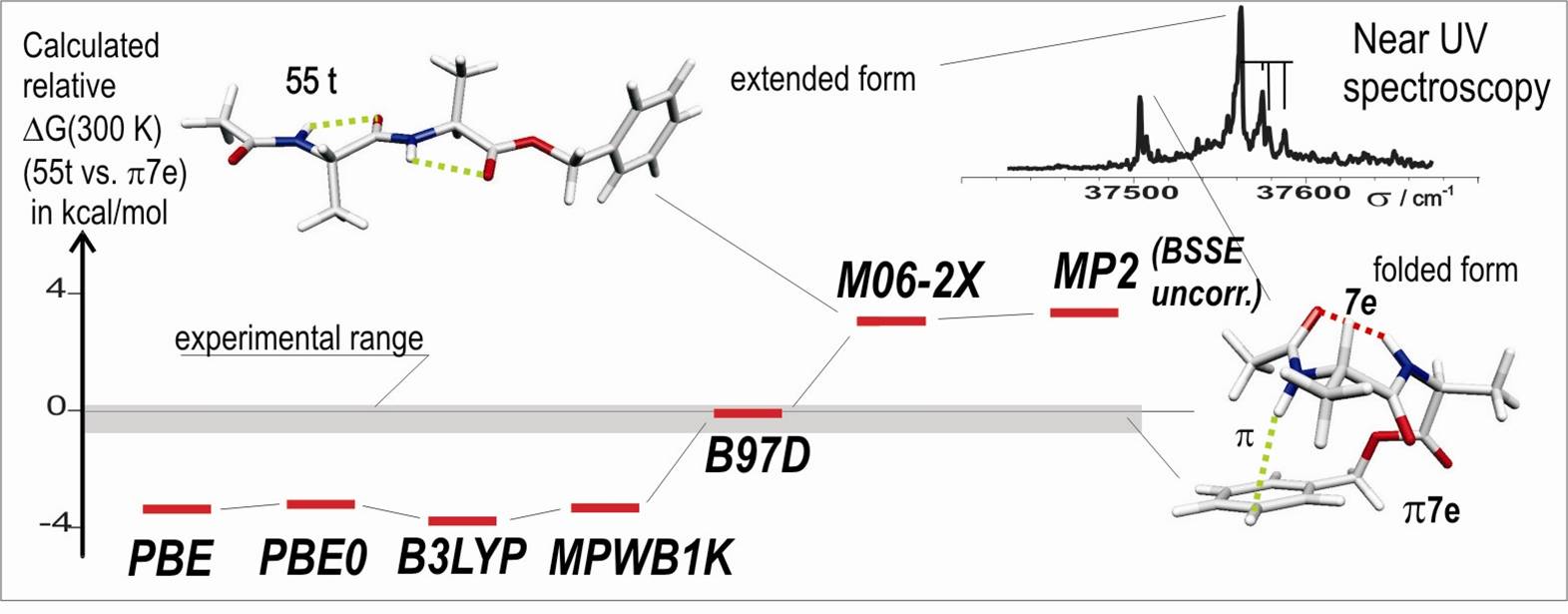
Ces données expérimentales, à savoir des énergies libres voisines pour ces deux espèces, ont permis d’évaluer les scores des méthodes actuelles les plus courantes de la chimie quantique. La figure montre l’énergie libre calculée pour la forme étendue par rapport à la forme repliée ; cette dernière présentant une importante composante dispersive stabilisante. Les méthodes DFT qui sous-estiment la dispersion prédisent une stabilité trop grande des formes étendues, incompatible avec l’observation des formes repliées. A contrario, les méthodes post-Hartre-Fock (MP2) ou des fonctionnelles récentes (M06-2X) élaborées pour prendre en compte la dispersion semblent la surestimer, conduisant à une surestimation de la stabilité de la forme repliée.
Ce "benchmarking" centré sur les interactions dispersives est crucial pour les chimistes "quanticiens", en leur proposant des systèmes tests auxquels confronter les méthodes qu’ils développent. Il sera poursuivi sur d’autres systèmes flexibles ainsi que sur les hydrates de ces molécules.
Référence :
Gas phase folding of a two-residue model peptide chain: on the importance of an interplay between experiment and theory,
E. Gloaguen, B. de Courcy, J.P. Piquemal, J. Pilmé, O. Parisel, R. Pollet, H.S. Biswal, F. Piuzzi, B. Tardivel, M. Broquier and M. Mons, J. Am. Chem. Soc. 13 (2010) 11860.
• ![]() Physique et chimie pour le vivant et l’environnement › Physique et vivant / Physics and life
Physique et chimie pour le vivant et l’environnement › Physique et vivant / Physics and life
• Laboratoire Interactions, Dynamiques et Lasers (LIDYL) - CEA-CNRS et Université Paris Saclay • Service des Photons Atomes et Molécules
• Laboratoire Francis Perrin • LUMO-SBM : Equipe Structures BioMoléculaires
