





|
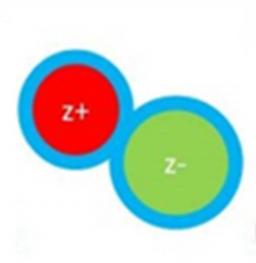
Les paires d’ions, atomiques ou moléculaires, sont naturellement omniprésentes, dans l’eau de mer, les aérosols et jusqu’au sein des organismes vivants. Elles s'assemblent dans les toutes premières étapes de la cristallisation des espèces ioniques, influencent les propriétés des solutions concentrées en ions ou des liquides ioniques, et jouent ainsi un rôle majeur dans de nombreuses applications. L'équipe SBM de l'UMR LIDYL a réalisé une première en isolant en phase gazeuse des paires d'ions complexes typiquement rencontrées en solution, et en les caractérisant par spectroscopie IR et UV. Cette approche expérimentale originale ouvre la voie vers une meilleure connaissance de ces objets supramoléculaires qui restaient non caractérisés individuellement jusqu'à présent. |
|
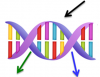
L'ADN de nos cellules est naturellement soumis à divers types de rayonnements. Parmi les plus énergétiques du spectre solaire, les ultra-violets sont susceptibles de provoquer de graves endommagements, et les mécanismes associés méritent d'être détaillés. L’énergie d’un photon absorbé par une double hélice d’ADN est redistribuée, au cours du temps, entre différents états électroniquement excités. La caractérisation de cette redistribution d'énergie est importante pour comprendre les processus d’endommagement de l’ADN par le rayonnement UV, mais est aussi utile pour la conception des matériaux bio-inspirés pour l’optoélectronique. Dans le cadre du projet ANR "DNAexciton", une collaboration internationale associée avec l'équipe du LIDYL a mis en évidence dans ce processus de relaxation la stabilisation d’un nouveau type d'excitons (ou paires électrons-trous) dénommés HELM (High-energy Emitting Long-lived Mixed states), avec des propriétés inattendues, tels que leur longue durée de vie. |
|
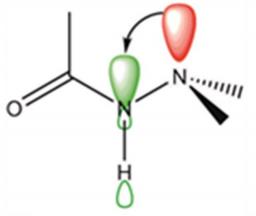
Les propriétés des molécules flexibles sont fonction de leur forme et leurs applications intéressent de multiples domaines (médicaments, nouveaux matériaux…). Un enjeu majeur dans ce domaine est de pouvoir contrôler leur repliement pour in fine contrôler leurs propriétés. De nombreuses techniques existent pour caractériser les différentes conformations que peuvent adopter ces molécules flexibles, dont la spectroscopie IR. La présente étude, impliquant une équipe du LIDyL en collaboration avec deux équipes de l’Université Paris-Sud, porte sur une fonction chimique très appréciée dans la synthèse de molécules flexibles de forme contrôlée : la fonction hydrazide (–CO-NH-N<). Les conformations des systèmes flexibles sont structurées en partie par l’hyperconjugaison (interaction stabilisante des électrons d’une liaison σ avec une orbitale vide adjacente) dont la mise en évidence expérimentale directe restait jusqu’à présent très limitée. Cette étude montre que la spectroscopie IR est extrêmement sensible aux effets d’hyperconjugaison se produisant au cœur des hydrazides, offrant ainsi un puissant diagnostic conformationnel. En parvenant à caractériser ces effets sur des hydrazides, la spectroscopie IR ouvre ainsi des perspectives non seulement pour la caractérisation des conformations de nombreux systèmes flexibles, mais aussi pour l’étude plus fondamentale du concept d’hyperconjugaison et de sa modélisation. |
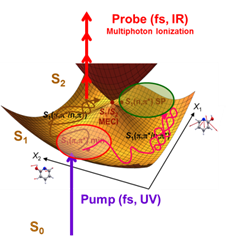
Elementary chemical acts are often associated with the crossing of a saddle point: 2 colliding reagents are climbing up a valley of the ground state potential energy surface which describes their interaction; they reach a saddle point and run downhill a valley toward the reaction products. The landscape is more complicated when electronic excitation is present since several potential surfaces are coupled, often via conical intersections (see Fig.1). Potential surfaces are very distorted in their vicinity, allowing ultra-fast exchanges - ~ 100 fs = 10-13 s or less - between electronic energy and deformation of the molecular skeleton. The Reaction Dynamics group at LIDyL/LFP is very active in this field for clarifying of the role of these intersections. It combines femtochemistry pump-probe experiments with sophisticated theoretical approaches.
A recent result on electronically excited 2-hydroxypyridine ![]() revealed a surprising dynamics (REF. and Fig.1). The molecule was excited in the S1 state (Fig.1, purple arrow), in the vicinity of a saddle point where the dominant electronic configuration is pp*. The wave packet which is formed moves out of this area along trajectories as those schemed as red and brown spaghettis in Fig.1. Departure from the excitation area takes place in ca 100 fs, mostly via deformations of the pyridine cycle. A slower presumably chaotic evolution follows (~1.3 ps) when the movement of the H-atom bonded to the O-atom couples other deformations of the molecular skeleton. This corresponds to a roaming behavior of the wave packet which spreads in areas where np* is the dominant electronic configuration. Roaming behaviors have been invoked repeatedly to interpret time unresolved experiments. The reference below reports for the first time their direct observation in a time-resolved femtochemistry experiment.
revealed a surprising dynamics (REF. and Fig.1). The molecule was excited in the S1 state (Fig.1, purple arrow), in the vicinity of a saddle point where the dominant electronic configuration is pp*. The wave packet which is formed moves out of this area along trajectories as those schemed as red and brown spaghettis in Fig.1. Departure from the excitation area takes place in ca 100 fs, mostly via deformations of the pyridine cycle. A slower presumably chaotic evolution follows (~1.3 ps) when the movement of the H-atom bonded to the O-atom couples other deformations of the molecular skeleton. This corresponds to a roaming behavior of the wave packet which spreads in areas where np* is the dominant electronic configuration. Roaming behaviors have been invoked repeatedly to interpret time unresolved experiments. The reference below reports for the first time their direct observation in a time-resolved femtochemistry experiment.
|
One of the goals in physical chemistry is to follow reaction paths in details. Many techniques have been developed with studies conducted in cells, effusive or supersonic beams, using laser spectroscopy, mass spectrometry etc. as investigation tools.
An original technique called "Cluster Isolated Chemical Reaction" (CICR) has been developed by the group "Dynamique Réactionnelle" of LIDyL/LFP for spectroscopic and reaction dynamics studies [1]. It allows determining the stoichiometry of the observed processes on a nanoreactor consisting of a van der Waals cluster. Recently, it has been successfully applied to helium droplet, which is a quite fascinating medium joining a very low temperature (0.37 K), superfluidity and finite size. The latter allows trapping and isolating a controlled number of molecules and stabilizing complexes which have a very low stability. The study of such systems provides very accurate information on the intramolecular potential energy surfaces (PES), improving the reliability of the associated models. |
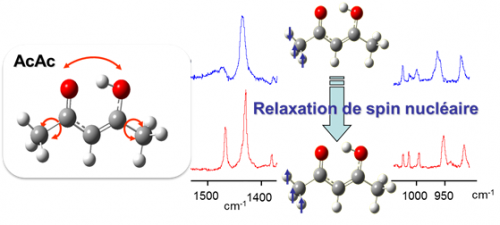
The stability and conformational dynamics of biomolecules are strongly influenced by the dynamics of hydrogen bonds and by hydrogen transfer processes which occur through these bonds. Not surprisingly, a large activity develops since many years to understand which molecular deformations play a role when a hydrogen atom is transferred through a hydrogen bond. The acetylacetone molecule (noted AcAc in the figure) is often used to approach this question from a fundamental point of view. Under the form drawn in the figure (the most stable one in the gas phase), AcAc is indeed one of the simplest molecule which carries an intramolecular hydrogen bond with the H-atom in proper position to transfer from an atom to another (here, the two oxygen atoms drawn in red in the figure). This issue relates to a more general one in reaction dynamics, a field in physical chemistry: to bring enough an accurate experimental material for modeling and foreseeing how several degrees of freedom couple together to drive to a chemical reaction.
The MOMA group of the Institute of the Molecular Sciences of Orsay (ISMO), in collaboration with the Reaction Dynamics group of the laboratory Francis Perrin (LFP) developed a very attractive method to show (maybe to control in the future) how large amplitude motions are coupled to frustrated rotations of the methyl groups in AcAc (schemed as bend arrows in the left panel of the figure) to stimulate the transfer of the H-atom between the two oxygen atoms (also shown by an arrow in the figure). This collaboration was initiated by the ANR project GOUTTELIUM and is pursued through the NOSTADYNE project funded by Triangle de la Physique. The AcAc molecule was isolated in a matrix of para-hydrogen, a quantum solid that does not perturb the deformation of hosted molecules. At the temperature of the matrix (4K), the relaxation of the nuclear spin of the methyl groups is very slow. This has been taken as an advantage to reveal that the pseudo-rotation of the methyl groups is intricate with the large amplitude motion associated with the H-atom transfer. The experimental method that lead to this result (see the reference) appeared as an elegant way, although indirect, to answer a long controversy on the role of the methyl rotations in the H-atom transfer in AcAc. The issue was important actually, since AcAc is considered as a prototype to unravel the dynamics of molecules possessing an intramolecular hydrogen bond.
|
By comparison with experimental data obtained specifically for this purpose by scientists of IRAMIS / SPAM, it is possible to select the best ab initio method able to give the parameters of "force field" models to faithfully reproduce the structure of assemblies of atoms, as complex as proteins (macromolecule composed of amino acids) or peptides (small polymers of amino acids, n <50). |
Clusters consists in a set of several up to million atoms in condensed phase with nanometric finite size. The investigation of their properties as the function of their increasing size is sometimes presented as one way to build a macroscopic condensed world from a gas. This often appears to be too simplistic, because the static and dynamic behavior of clusters has often no equivalence at the macroscopic scale, as they are controlled by their high specific surface and their finite size. These concerns the physical chemistry of the mesoscopic world, as illustrated by the recent experimental study driven by the "Reaction dynamics" group of DRECAM/SPAM-Laboratoire Francis Perrin in collaboration with a theoretician team from Paris VI University.

