










Environments opens intermolecular relaxation channels. The goal of this study is to investigate the competition between intermolecular and intramolecular relaxation processes. It goes through the study of atoms, Rydberg molecules, etc...
Dynamics of highly excited barium atoms deposited on large argon clusters. I. General trends
A. Masson, L. Poisson, M. A. Gaveau, B. Soep, J. M. Mestdagh, V. Mazet and F. Spiegelman
J. Chem. Phys., 133, 054307, 2010 [doi]
Ba(Ar)(approximate to 750) clusters were generated by associating the supersonic expansion and the pick-up techniques. A femtosecond pump (266.3 nm)-probe (792 or 399.2 nm) experiment was performed to document the dynamics of electronically excited barium within the very multidimensional environment of the argon cluster. Barium was excited in the vicinity of the 6s9p P-1 state and probed by ionization. The velocity imaging technique was used to monitor the energy distribution of photoelectrons and photoions as a function of the delay time between the pump and the probe pulses. A complex dynamics was revealed, which can be interpreted as a sequence/superposition of elementary processes, one of which is the ejection of barium out of the cluster. The latter has an efficiency, which starts increasing 5 ps after the pump pulse, the largest ejection probability being at 10 ps. The ejection process lasts at a very long time, up to 60 ps. A competing process is the partial solvation of barium in low lying electronic states. Both processes are preceded by a complex electronic relaxation, which is not fully unraveled here, the present paper being the first one in a series. (C) 2010 American Institute of Physics. [doi:10.1063/1.3464489]
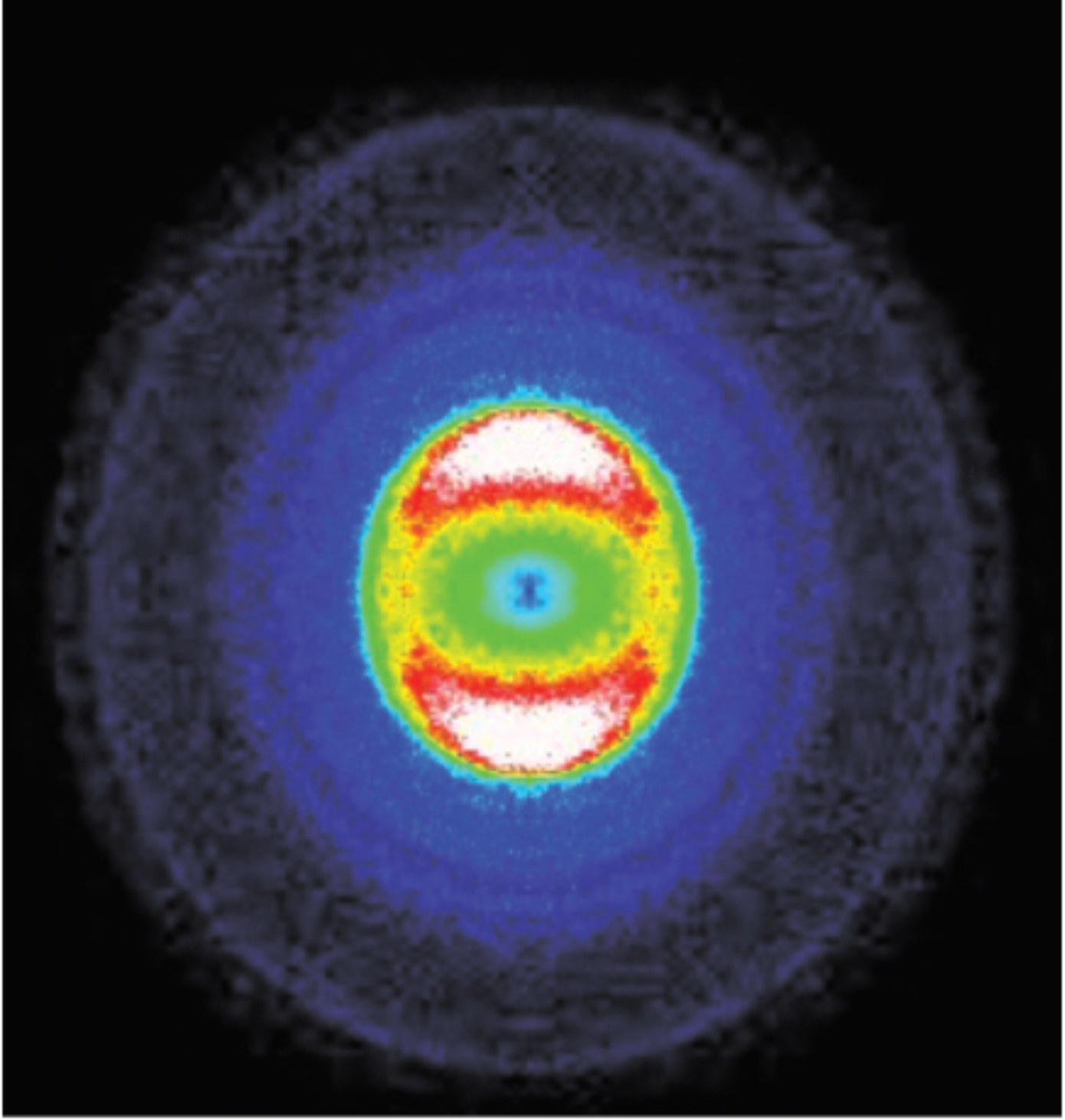
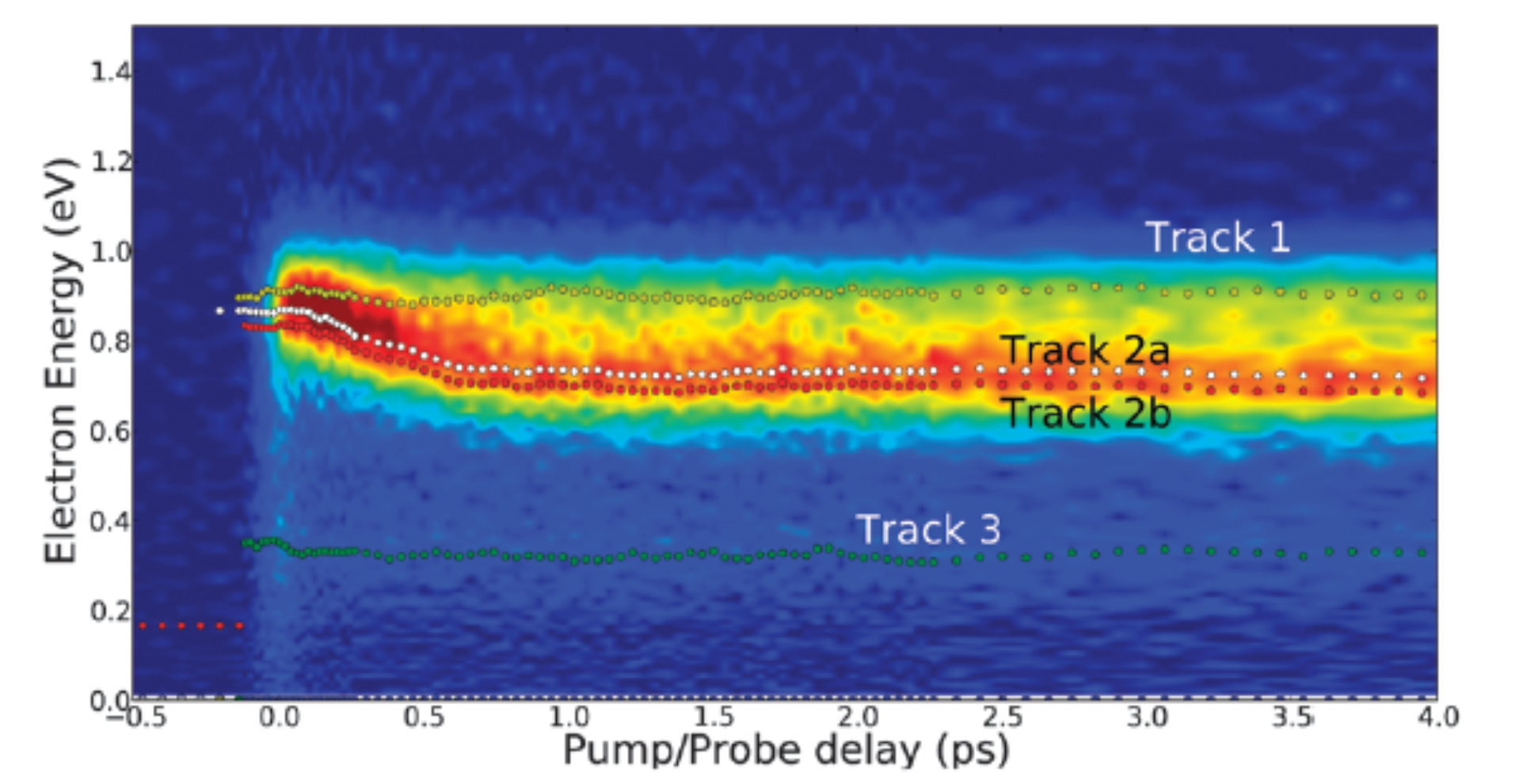
Time resolved observation of the solvation dynamics of a Rydberg excited molecule deposited on an argon cluster-I: DABCO* at short times
S. Awali, L. Poisson, B. Soep, M.-A. Gaveau, M. Briant, C. Pothier, J.-M. Mestdagh, M. B. E. H. Rhouma, M. Hochlaf, V. Mazet and S. Faisan
Phys. Chem. Chem. Phys., 16, 516, 2014 [doi]
This paper is a joint experimental and theoretical approach concerning a molecule deposited on a large argon cluster. The spectroscopy and the dynamics of the deposited molecule are measured using the photoelectron spectroscopy. The absorption spectrum of the deposited molecule shows two solvation sites populated in the ground state. The combined dynamics reveals that the population ratio of the two sites is reversed when the molecule is electronically excited. This work provides the timescale of the corresponding solvation dynamics. Theoretical calculation supports the interpretation. More generally, close examination of the short time dynamics (0-6 ps) of DABCOArn gives insights into the ultrafast relaxation dynamics of molecules deposited at interfaces and provides hence the time scale for deposited molecules to adapt to their neighborhoods.
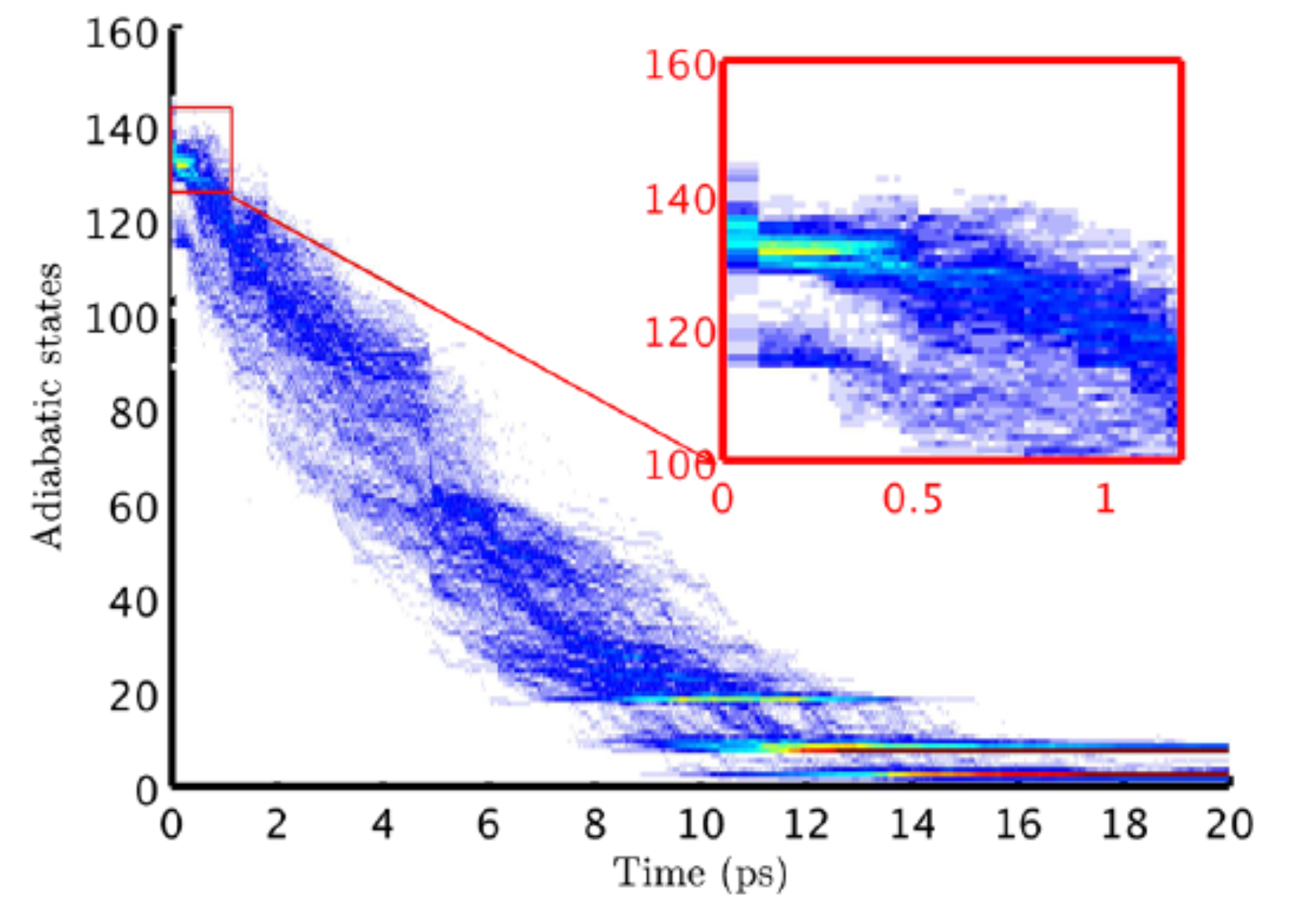
Coupled Electronic and Structural Relaxation Pathways in the Postexcitation Dynamics of Rydberg States of BaArN Clusters
A. Masson, M. C. Heitz, J. M. Mestdagh, M. A. Gaveau, L. Poisson and F. Spiegelman
Phys. Rev. Lett., 113, 123005,2014 [doi]
We investigate, theoretically, the joint relaxation of orbital and structure in postexcitation dynamics of Rydberg states of cluster BaArN (N = 250). Mixed quantum-classical dynamics is used to account for the nonadiabatic transitions among more than 160 electronic states, represented via a diatomics-in-molecules Hamiltonian. The simulation illustrates the complex multistep relaxation processes and provides detailed insight in the mechanisms contributing to the final-time experimental photoelectron spectrum.
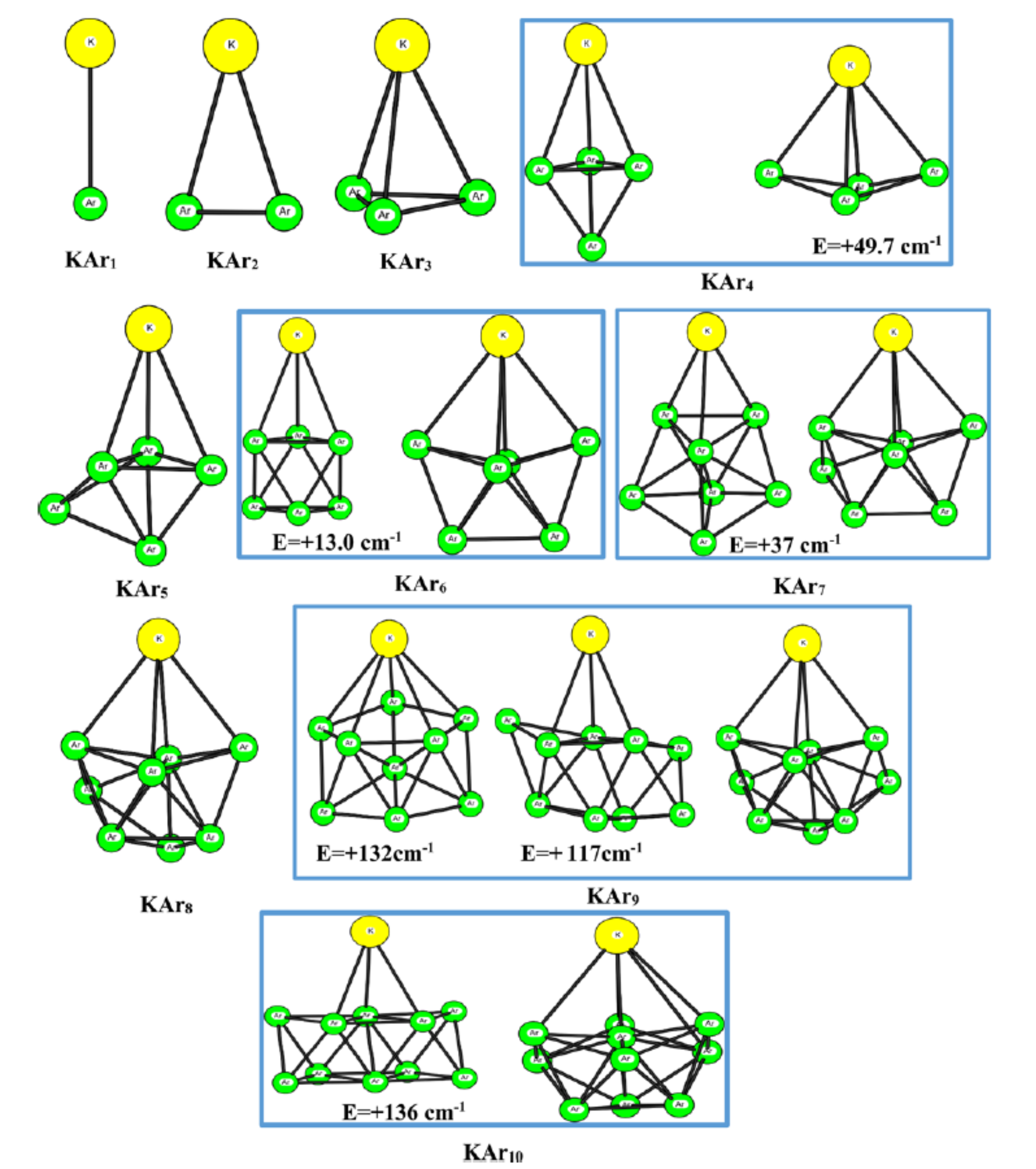
Absorption Spectroscopy, a Tool for Probing Local Structures and the Onset of Large-Amplitude Motions in Small KAr(n) Clusters at Increasing Temperatures
S. Awali, L. Poisson, M. B. E. H. Rhouma and J.-M. Mestdagh
The journal of physical chemistry. A, 119, 9729, 2015 [doi]
Photoabsorption spectra of KArn (n = 1-10) are simulated at temperatures ranging between 5 and 25 K. The calculations associate a Monte Carlo (MC) method to sample cluster geometries at temperature T, with a one-electron ab initio model to calculate the ground-state and excited-state energies of the cluster. The latter model replaces the K(+) core electrons and all the electrons of the Ar atoms by appropriate pseudopotentials, complemented by core polarization potentials. It also provides the necessary oscillator strengths to simulate the spectra. Global optimization by basin-hopping is used in combination with MC simulation at low temperature (5 K) to identify the most stable isomer and remarkable isomers of ground-state KArn clusters, which are stable with respect to deformations of the order of those expected with Zero Point Energy motions. The absorption spectra calculated for each of these isomers at 5 K suggest that absorption spectroscopy can probe sensitively the local environment of K atom: surface location of K with respect to a close-packed Ar moiety, number of Ar atom in close vicinity, and local symmetry about K. Simulation at increasing temperatures, up to the evaporation limit of K out of the cluster, shows the onset of large amplitude motions above 20 K, when the K atom experiences a variety of local environments.
Spectroscopy and Dynamics of K Atoms on Argon Clusters
J. Douady, S. Awali, L. Poisson, B. Soep, J. M. Mestdagh and B. Gervais
J. Phys. Chem. A, 119, 6074, 2015 [doi]
We present a combined experimental and simulation study of the 4s -> 4p photoexcitation of the K atom trapped at the surface of ArN clusters made of a few hundred Ar atoms. Our experimental method based on photoelectron spectroscopy allows us to firmly establish that one single K atom is trapped at the surface of the cluster. The absorption spectrum is characterized by the splitting of the atomic absorption line into two broad bands, a Pi band associated with p orbitals parallel to the cluster surface and a Sigma band associated with the perpendicular orientation. The spectrum is consistent with observations reported for K atoms trapped on lighter inert gas clusters, but the splitting between the Pi and Sigma bands is significantly larger. We show that a large amount of K atoms are transiently stuck and eventually lost by the Ar cluster, in contrast with previous observations reported for alkaline earth metal systems. The excitation in the Sigma band leads systematically to the ejection of the K atom from the Ar cluster. On the contrary, excitation in the Pi band leads to the formation of a bound state. In this case, the analysis of the experimental photoelectron spectrum by means of nonadiabatic molecular dynamics simulation shows that the relaxation drives the system toward a basin where the coordination of the K atom is 2.2 Ar atoms on the average, in a poorly structured surface.
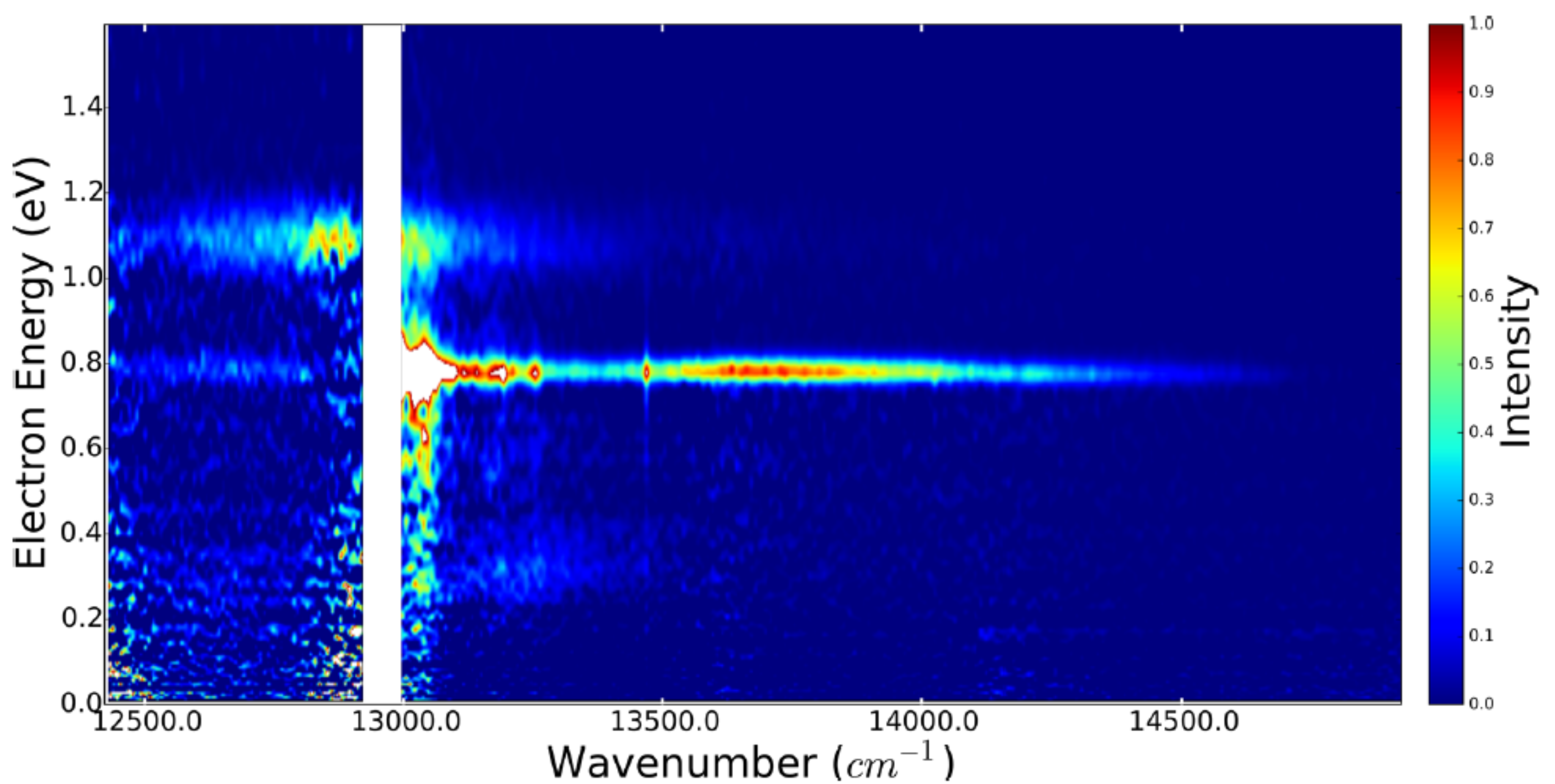
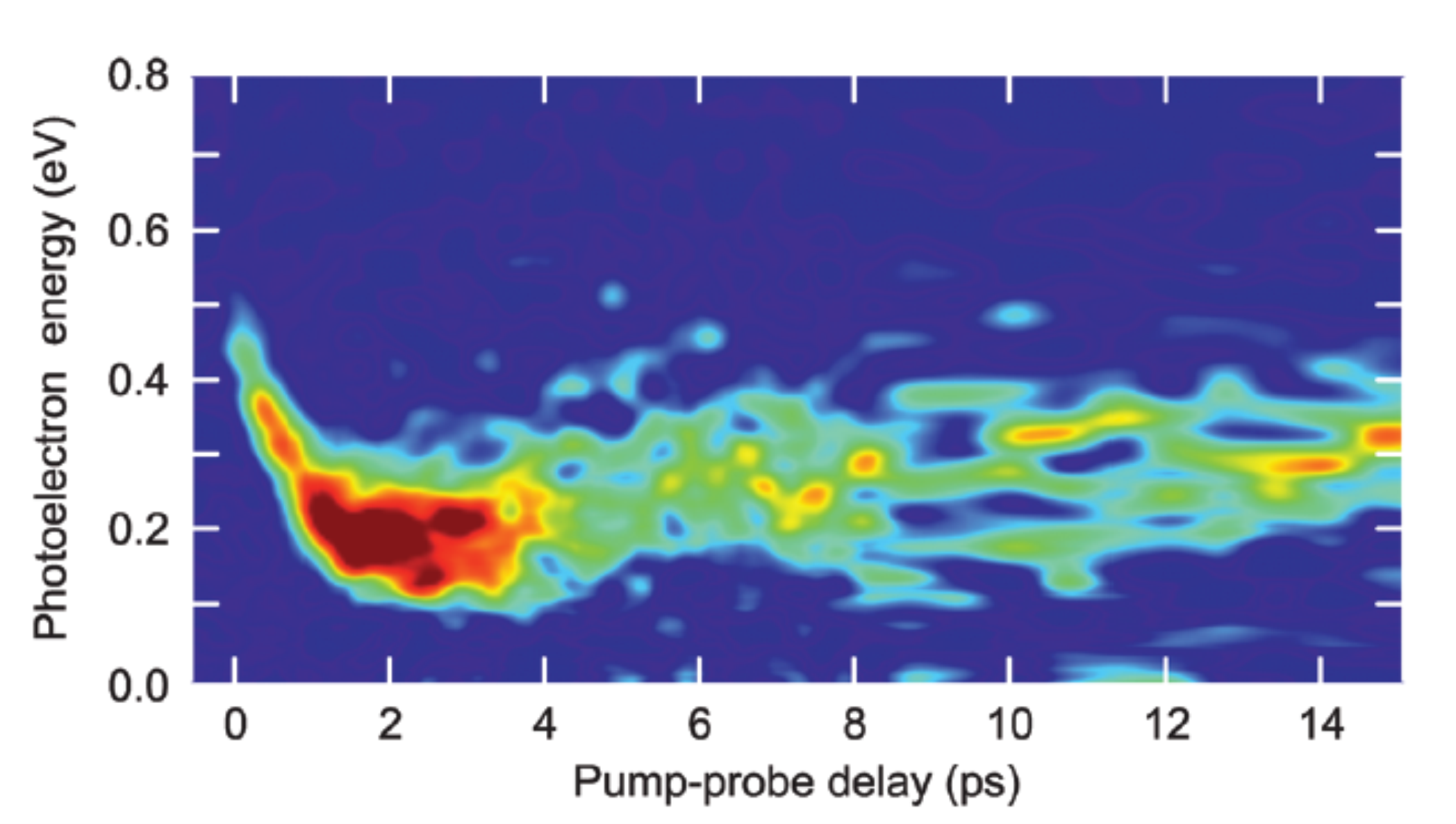
Multipronged mapping to the dynamics of a barium atom deposited on argon clusters
S. Awali, M. A. Gaveau, M. Briant, J. M. Mestdagh, B. Soep, O. Gobert, R. Maksimenka and L. Poisson
Phys. Chem. Chem. Phys., 18, 32378 2016 [doi]
The dynamics of an electronically excited barium atom deposited at the surface of an Ar500 cluster was explored in a multipronged approach which associates information from frequency-resolved nanosecond experiments and information from femtosecond time-resolved experiments. In both types of experiments, the dynamics is monitored by photoelectron and photoion spectroscopy.
• ![]() Interaction laser-matière › Physico-Chimie et Chimie-Physique
Interaction laser-matière › Physico-Chimie et Chimie-Physique
• Laboratoire Interactions, Dynamiques et Lasers (LIDYL) - CEA-CNRS et Université Paris Saclay
• LUMO-DyR - Reaction Dynamics Team • LUMO - Equipe Dynamique Réactionnelle (DyR)
