









Les activités théoriques concernent le traitement par des méthodes de chimie quantique de systèmes moléculaires de grande taille, un challenge théorique pour ces méthodes. Elles sont réalisées, soit en lien direct avec les expériences en phase gazeuse menées au sein du groupe sur les systèmes d’intérêt biologiques, soit dans le cadre des missions nucléaires du CEA. Les récents développements des méthodes de chimie quantique sont donc utilisés afin de mettre en œuvre des stratégies de calculs et/ou des modèles théoriques permettant d’obtenir une meilleure compréhension de ces systèmes complexes.
Les systèmes d’intérêt biologique sont des systèmes très flexibles, de taille moyenne (10–100 atomes), dont les structures sont régis par des interactions non-covalentes et dont les états excités de nature très diverses (états localement excités et états à transfert de charge).
La modélisation de leur paysage conformationnel à l’état fondamental est donc basée sur une stratégie théorique qui comporte plusieurs étapes avec in fine une confrontation avec l’expérience pour l’assignement des structures observées :
- (i) exploration de la surface d’énergie potentielle par des méthodes globales d’exploration couplée à différents champs de forces
- (ii) optimisation de la géométrie des conformations sélectionnées par une méthode de chimie quantique appropriée (DFT-D, Théorie de la fonctionnelle de la densité avec un terme semi-empirique supplémentaire pour la dispersion) et
- (iii) simulation des spectres IR dans l’approximation harmonique avec correction par des facteurs d’échelle mode-dépendants.
La modélisation de la dynamique des états excités, elle, comporte deux volets :
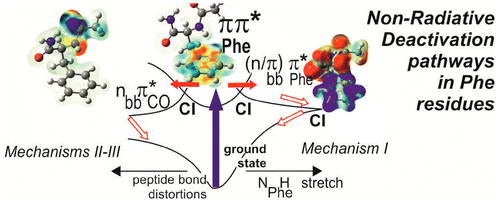
- (i) l’énergétique des états excités des résidus aromatiques (notamment la Phénylalanine) dans des environnements différents et
- (ii) la dynamique des premiers états excités par dynamique non-adiabatique (Formalisme de la DFT dépendante du temps -TDDFT) afin de cerner le rôle des intersections coniques et de déterminer les coordonnées pertinentes des mécanismes de relaxation (Coll. Institut Rudger Boskovic, Zagreb, Croatie) suivie d’une caractérisation plus précise des surfaces de potentiel par une méthode de chimie quantique sophistiquée (Méthode Coupled Cluster à l’ordre 2, CC2). Cette caractérisation sera prochainement confortée par l’utilisation d’une méthode d’interaction de configuration multiréference utilisant des orbitales localisées (CAS-SDCI, Coll. LCPQ, Toulouse) en des points remarquables des surfaces d’énergie potentielle, comme les minima, les états de transition et bien sur les intersections coniques.
La modélisation des matériaux combustibles sous irradiation a pour objectif une meilleure compréhension des mécanismes élémentaires gouvernant les propriétés des matériaux nucléaires afin d’améliorer la précision des codes de performances des combustibles. Elle concerne notamment la validation de nouvelles approches de la DFT qui permettent a priori de traiter correctement les forces dispersives, forces responsables de nombreux phénomènes structuraux et énergétiques engendrés dans les matériaux sous irradiation comme la création de défauts ponctuels et étendus, la production d’hélium et de fragments de fission, dont des gaz rares. Ces études ciblent pour l’instant les interactions entre les gaz rares et des atomes à couche électronique incomplète dans des systèmes modèles contenant des petites molécules. Elles visent à être étendues à des modèles plus réalistes, i.e. des petits agrégats de carbure de silicium (SIC) représentant 1 ou 2 couches de coordination du gaz rare, SIC étant un matériau d’intérêt dans le domaine du nucléaire (Coll. CEA - DEN/DEC/SESC/LCC et Projet Fédérateur NEEDS FP-Matériaux). Enfin, elles sont toujours réalisées par confrontation des résultats obtenus en DFT avec ceux donnés par une des méthodes ab initio les plus sophistiquées, la méthode Coupled Cluster simple-double avec correction des triples (CCSD(T)).
