

The theoretical activities are focussed onto the modelling of large molecular systems using quantum chemistry methods, which is a theoretical challenge for these methods. These modeeling actions are either connected to the gas phase experimental investigations carried out in the group, or are done in support of nuclear activities of the applied programs of CEA. Recent developments in quantum chemistry methods are thus used to implement strategies of calculations and / or theoretical models to obtain a better understanding of these complex systems.
The systems of biological interest are very flexible systems, medium-sized (10-100 atoms) systems, whose structure is controlled by non-covalent interactions and whose excited states can exhibit very different natures (e.g., locally excited states, charge-transfer states, etc...).
Modelling their ground state conformational landscape is based on a multistep theoretical strategy, leading in fine to a confrontation with the experimental data:
- i) an exploration of the potential energy surface using global exploration methods coupled to various force fields,
- ii) a geometry optimisation<:strong> of a selected set of conformations using a convenient quantum chemistry method (DFT-D, Dispersion-corrected Density Functional Theory)
- (iii) calculation of the IR spectra in the harmonic approximation and with mode-dependent scaling factors.
The excited state modelling includes two actions:
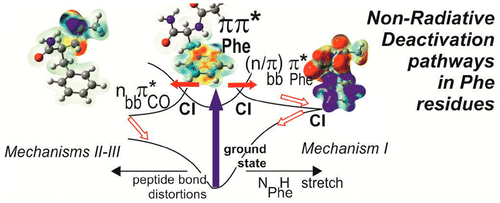
- i) the excited state energetics of aromatic residues (namely Phenylalanine) in various environments, and
- ii) the non-adiabatic dynamics of the first excited states (using the Time-Dependent Density Functional Theory, TD-DFT, formalism) in order to assess the role of conical interesections between these states and to determine the critical distortions responsible for the relaxation (in coll. with the Rudger Boskovic Institute, Zagreb, Croatia). This is followed by a more precise characterization of the potential energy surfaces using a more accurate quantum chemistry method (2nd order Coupled-Cluster - CC2- method). This characterization will be confirmed in a near future by using a multirefrence configuration interaction method using localized orbitals (CAS-SDCI, Coll. LCPQ, Toulouse) in order to calculate energetics of remarkable points of the surface, like the minima, the transition states and of course the conical intersections.
Atomic scale modelling of Nuclear Fuel Materials under irradiation aims at a better understanding of the elementary mechanisms controlling the properties of nuclear materials in order to improve the precision of the models describing the nuclear fuel characteristics. It involves in particular the validation of new DFT approaches in order to describe correctly dispersive interactions, which is mandatory to account for several structural and energetic phenomena taking place in materials under irradiation, namely the creation of defaults, localized or extended, or the interaction of helium atoms as well as other atomic fission products, like rare gases, with the materials.
These studies are focussed onto the interaction or rare gases with open shell atoms in small model systems containing small molecules, and will be extended to more realistic models, namely small SiC clusters, representing one or two solvation shells of a rare gas atom; SiC being a typical material of interest for nuclear industry (Coll. CEA - DEN/DEC/SESC/LCC et Projet Fédérateur NEEDS FP-Matériaux). These DFT calculations are systematically validated by comparison with the more accurate quantum chemistry method, i.e., Coupled Cluster at the CCSD(T) level.
