... Phases Magazine N° 20
Transport électronique à travers un
atome
SEPTEMBRE 1999 N° 21
|
|
Une nouvelle imagerie des cristaux à partir de données de diffraction des rayons X et des neutrons est développée au Laboratoire Léon Brillouin (LLB) depuis 1990. Elle utilise un outil mathématique qui a déjà fait ses preuves en astrophysique et en médecine, qui s'appuie sur le principe du maximum d'entropie.
On peut citer de nombreux problèmes de structure des matériaux encore mal résolus. Par exemple, comment se comporte une molécule de symétrie donnée placée sur un site cristallin de symétrie différente ? Où se trouvent les atomes d'oxygène interstitiels dans un catalyseur ou dans un oxyde supraconducteur ? Où se trouvent les sites actifs dans les cavités d'un matériau poreux comme une zéolite ? Expérimentalement, si la diffusion inélastique des neutrons est particulièrement adaptée à l'étude spectroscopique des protons dans les solides, elle présuppose une bonne connaissance préalable des propriétés structurales des composés étudiés. Par ailleurs, des calculs de modélisation moléculaire dans les solides prédisent les comportements structuraux et dynamiques pour les atomes ou les molécules, mais ils requièrent néanmoins la détermination expérimentale précise des propriétés structurales pour la validation de leurs résultats.
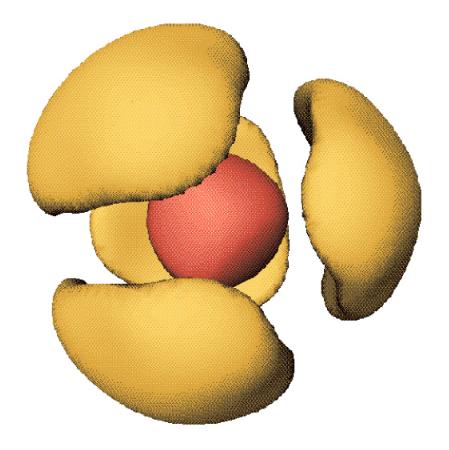
Notre objectif est de trouver la "meilleure" densité tridimensionnelle électronique [cas des rayons X] ou nucléaire [cas des neutrons non polarisés] ou encore d'aimantation [cas des neutrons polarisés]. Cette densité doit permettre d'ajuster au mieux les mesures d'un ensemble limité de composantes de Fourier, toujours bruitées et parfois groupées et indissociables. Le plus souvent, l'analyse conventionnelle des spectres de diffraction, basée sur les synthèses de Fourier, ne permet pas d'extraire l'information structurale recherchée avec une précision suffisante (la fraction d'Angström ; 10-10 m). Cette motivation nous a conduit à développer une imagerie performante qui appliquée aux données de diffraction permet de discerner individuellement atomes, molécules et spins (Fig. 1). Cette méthode, qui a fait ses preuves pour les études réalisées sur monocristaux, s'applique également pour les mesures sur poudre, en particulier lorsque l'expérience est réalisée sur un synchrotron car on peut alors obtenir des données d'une très grande résolution.
|
|
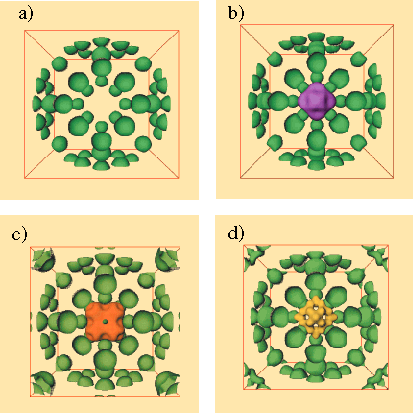
On cherche ici à localiser une molécule hôte (1,3 dioxalane) dans une cage de zéolite. Un tel système est seulement disponible sous forme de poudre. Les intensités des pics de diffraction de rayons X mesurés, ont été utilisés pour obtenir les modules des composantes de Fourier de la densité électronique cherchée. Les phases correspondantes sont inconnues, mais indispensables à la détermination complète de la structure. Ici, elles sont calculées à partir d'un modèle structural a priori décrivant la cage vide de la zéolite.
La reconstruction est alors objective vis à vis de la partie inconnue, la molécule hôte. Les composantes phasées de la cage seule [généralement connue, de type sodalite dans le cas de la figure 2] sont tout d'abord calculées et arbitrairement bruitées. La cage vide (Fig. 2a) sert de référence ("phantom"). Sa reconstruction à partir de données simulées est utilisée pour fixer les limites de fiabilité de la reconstruction dans le cas de données réelles. Trois autres reconstructions structurales sont alors effectuées à partir des données expérimentales. Une reconstruction utilisant les méthodes standards (Fourier différence, Fig. 2b) sert de noyau d'évaluation de la nouvelle analyse proposée. La basse résolution obtenue exclut l'observation de tout détail. Ensuite, une reconstruction entropique naïve est réalisée sans utiliser la connaissance que l'on a de la cage vide. Enfin, une reconstruction entropique optimisée qui s'apparente à une Fourier différence entropique, fournit la densité électronique cherchée. C'est celle qui reconstruit le mieux possible la cage vide puis utilise le reste de l'information expérimentale disponible pour reconstruire la partie inconnue. Les détails fins qui apparaissent ainsi sur la Fig. 2d peuvent être interpréter comme des sites préférentiellement occupés par les atomes de la molécule hôte (ici, de 1,3 dioxalane). Un modèle pour le comportement de celle-ci est alors construit, qui est compatible avec ses propriétés physiques lorsqu'elle est isolée. Le modèle est ensuite validé si il apporte une amélioration sensible du facteur d'ajustement des données calculées aux données expérimentales.
L'étape suivante de notre travail, actuellement en cours, consiste à adapter cette méthode pour l'appliquer sur des données de diffraction de neutrons sur poudre acquises au LLB en collaboration avec J. Rodriguez-Carvajal. La difficulté supplémentaire provient alors du fait que la résolution instrumentale est d'un ordre de grandeur moins bonne que dans le cas de mesures par diffraction de rayons X.
Pour en savoir plus :
R.J. Papoular ''Cristallographie et Reconstruction d'Images par
Maximum d'entropie''
Rapport CEA-R5758 (1997)
K. Knorr et al ''Model-free reconstruction of host/ guest compounds from powder
diffraction data''
Microporous & Mesoporous Materials 21 (1998) 353-363
S. Webb ''The Physics of Medical Imaging''
S. Webb ed, Ch12, Institute of Physics (1998)
Contact :
R. Papoular.
Phases Magazine N° 21
Des harmoniques plus vives que l'éclair…